This spring we organized the KTH course Scientific Programming in Python for Computational Biology, hosted at SciLifeLab Stockholm.
The course ran over seven weeks with one two hour session every wednesday, with alotted time after the session for
students to ask questions about assignments. The examination was by passing the weekly assignments in a timely fashion.
Our twist on this classic concept was that all hand ins had to be done via Github.
The structure of the course was inspired by Software Carpentry.
There was demand for a course that goes beyond the basics of Python, and one challange with this is that even thought
there are plenty of online resources for introducing people to Python, structured lectures, and in particular exercises,
of more advanced sections are hard to come by.
We had a brainstorming session where we talked about what kind of things we wanted to cover in the course.
The course was intended for scientists, but in the spirit of software carpentry, we also wanted to teach good
software engineering practices.
A survey to various mailing lists to gage the interest of a Python course for scientists and how they would
feel about a more advanced course rather than the normal introductory courses.
Responses for the survey were great, though most people had stated they would prefer a simpler course, we felt
that those are already available several times per year. When we announced the course we asked people who intended to
attend to sign up on a mailing list. We got some 100 people signed up, but we figured plenty of people signed up for
the list simply because it was easy, and wasn’t really planning to attend. However, when the course started, we had
roughly 70 people on the first session! We had booked the largest presentation room in the building, but it was filled
to the brim. Personally I had expected the course to be more like a 10-15 people seminar. Now it was something else.
Everything was in Github
In the first session, Roman Valls Guimera gave an interactive lecture where students were to set up their Github accounts,
join the Github Organization for the course, and learn how to clone their repos for the course to their computers.
Among many other things he taught the students how to structure a Python project. The thought was that at the end,
the students should all have a repository for the course which we could later build upon with examples.
Part of the first assignment was to have everything set up so that you could push code to your personal repository
in the course organization. Every student had a repository named after their last name. Surprisingly, there were no
overlapping last names!
There were a couple of points of confusion here. Some inconsistencies in examples, along with a confusion even among
us organizers about where the repo’s for the course should be kept, caused students repositories to be quite varied.
But after just this first session we were lenient in the correction, enjoying the success of getting over 50 people
to use git for the first time and join Github!
The takeaway from this in the end, is that consistency of organization and examples would have helped the students
understand things quicker. And we organizers definitaly could have done a better job communicating with each other in
the beginning.
A key here is that we forced the students to use version control, and while we didn’t go through anything particularly
advanced, not even branching, getting people to do commits and push to a central server habitually is something that
courses normally don’t do.
IPython Notebooks rather than slides
In the second session I talked about how to parse various file formats with Python, and gave recommendations
for packages to use in various occasions. Actually, I can show exactly what I talked about, because for rather
than using slides as is common, live coding, I used IPython notebooks which I kept on Github for students to read
after the sessions.
Session 2 - Getting Data
Personally, I think the notebook is optimal for teaching Python. It takes a bit of explaining what it actually is, and
how it works. (That you can run code in cells of a browser is not intuitive when you’re just getting used to the idea
of running code in the terminal, as was the case for many students.)
It enables you to prepare a lecture in advance, like slides, but also gives you the abality to change things, or quickly
show things you didn’t think of when students have questions.
The session also included a guest lecture by Mats Dahlberg showing how to talk to relational databases with Python in
the second hour.
At the end, I went through how to talk to web services using requests. This is a bit unconventional, but we have the
convention at SciLifeLab to write and use RESTful API’s, and then it makes a lot of sense to show people how to actually
use these.
For the assignment the students were supposed to fetch some data from the web and do some calculation. But as a part
of this they were also supposed to structure the repo, and make setup.py install scripts so they could be used
globally.
Originally, I wanted to use data from the public transport network about the bus stop outside the SciLifeLab building,
but it turned out that data was not available without a for-pay API key. So instead I used the New York City transit
system data as an example. An idea I got from Python for Data Analysis.
To push the students to structure their repositories the way that was originally intended I explicitly wrote out
what kind of structure they should have after the assignment was done:
[lastname]/
[lastname]/
__init__.py
scripts/
getting_data.py
README.md
setup.py
This caused some to change their structure at least. Since we had so many students, we really wanted the repos to look
basically the same, so we could easily correct assignments, and assist students that needed help without having to
figure out their repo structure.
In the end, we never required the students to use the repos in the organization account, and allowed them to update the
fork of the repo they had on their personal accounts in stead. Thinking back, it would have been much better to only
look at the repos in the organization.
Self correcting assignments
The third session was about some modern Python idioms that people normally don’t see in an introductory course. You can
see the notebooks online.
The session also included a brief introduction to NumPy, and I remember talking a fair amount about the difference
between lists and arrays.
Session 3 - Modern Python Idioms
When preparing the session I was struggling with three things.
- I wanted a self contained example that would use what I had talked aboud.
- Many students still had inconsistant repository structures.
- I wanted to find time to write a script that would correct assignments automatically.
Out of all the nice things we did with the course, this is the one I’m the proudest of, I made the assignment so that
the students were supposed to make a class who’s objects would be used to check that any course repository was correct.
This is an assignment that will correct itself! A script was supposed to be written which would output the
string “PASS” is the specification of the directory was satisfied, and “FAIL” otherwise.
I made sure to be very explicit in what kind of structure the RepoChecker class should check for, and pointed the
students to use git mv if their own repos didn’t satisfy the structure the checker was looking for.
After this session almost every student had the required structure on their repos!
Use data about students generated through the course
The fourth session was much about not reinvinting the wheel, in particular about data containers. I mostly went through
use cases for the more useful types in the built in collections module.
Session 4 - Don’t reinvent the wheel
I also talked about SciPy and the SciKits. I didn’t want to delve too deeply in to any particular SciPy submodule
as the course was supposed to be wide, and honestly, for bioinformaticians SciPy doesn’t contain extremely useful
submodules.
Something I was glad to show and go through was Pandas, a package I really enjoy using. One thing to note, is that
though the rendering of a DataFrame as a table in the IPython notebook is really nice, some students got confused of
whether Pandas worked in the terminal, so I had to open a terminal and show a couple of examples.
Data containers are used everywhere, but the different ones are used in rather particular instances. So I didn’t even
try to think of an assignment that would use the ones I talked about. In stead, I wanted the students to do an assignment
where they used Pandas.
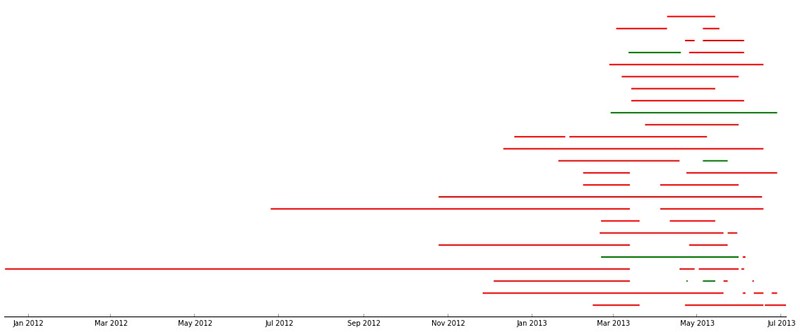
Inspired by the previous weeks assignment, I thought the data could be self generated as well. As the course
had been going on for a few weeks now, there was quite a bit of activity on the Github repositories. You can see
the entire formulation of the assignment at the end of the linked notebook. (I was even more explicit in the
description this time. Really, if you want to be able to correct things automatically, you need to be very explicit.)
In gist, the assignment was to use the RESTful API of Github to take data about commits to the organization repos
and put this in a Pandas DataFrame of time series, which they then couls use to find trends in the data.
Result: All students found that the vast majority of commits occured the hour before the weekly session (when also
the assignment deadline occured).
Many of the students had problems with this assignment, in particular due to one repository that ruined most people
strategies for fetching the data. In retrospect I think this was actually a good thing, it really demonstrated how
programming turns out a lot of the time in the real world, they had to figure out how to handle the special case.
Scientists should learn CI and testing
In the fifth session Roman returned, and talked about how to work with tests, and how to use a continous integration
system. He actually blogged about this here: Automated Python education via unit testing and Travis-CI
Rather than writing too much about it I’ll encourage anyone interested to read his blog post.
The session also had Hossein Farahani showing the graph package Networkx.
(Try to) use the code the students created.
For the sixth session I talked about how to optimize Python code, and quite in depth about how to use iterators /
generators.
Session 6 - Optimizing Python
This also lead to an interesting construct in Python that I see very few people use, the coroutines. These are in essence
the reverse of a generator, have a look at the notebook if you’re interested.
At the end I talked about how to use various profilers. When preparing this part of the lecture I realized even more
how handy the IPython notebook is for teaching. By using the %%bash cell magic, I could store bash commands, and
run them and see the output in a very nice way. In the process I also learned about the ability of starting background
processes in a notebook cell using %%script --bg bash.
For the assignment, again thinking that it’s the best for the students to work with what they have already created, I
figured they could profile the scripts they had made and report the results (by saving the outputs in the repo).
This, however, did not work out at all. (Yes this means the headline was never achieved). One of the profilers I
wanted them to use turned out to be more experimental than I had thought, and just plain crashed on the students code.
Various other differences in how the students had structured their scripts caused them to not be able to run several of
the profilers.
In addition to this, their script weren’t doing enough job for the profilers to have much time finding anything
interesting.
In the end, we had to give the students an extra week to complete the assignment. And in stead of them using their
scripts I found a script online for factoring numbers using the Elliptic Curve Method. I made sure to give very explicit
instructions, and made sure the profiling worked.
Parallelization is hard but rewarding
For the seventh and final session of the course I went through the two most common strategies for parallelization.
The multiprocessing module for programmatic parallelization, and IPython.parallel for interactive explorative
parallelization.
Session 7 - Parallel Python
This session was very popular, it seemed a lot of people realized many of their time consuming problems could be
rather easily parallelized to cut down computational time significally. As far as the lecture went, I just described
how to use the various components of the modules. For this session the students had plenty of questions, and the
feedback about the notebook was that it should have contained more comments and explanatory text.
One particular thing, is that multiprocessing doesn’t work in interactive prompts, so I had to make use of the
%%file cell magic to write tiny scripts for each example, then again use the %%bash magic to run them. It went
rather smooth, and I think the students understood what was going on.
Another detail, is that I piped the interactive notebook from an 8 core desktop to my laptop, as I figured the
examples would be quite dull with my dual core laptop.
For the assignment I wanted the students to factorize many numbers, and calculate some statistics about the factors
afterwards. So that this would be emberrasingly parallel over the list of numbers to factor. I wanted them to
implement this using both multiprocessing and IPython.parallal.
It turned out that IPython.parallel was very bad for this problem, and everyone was confused as to why the parallel
implementation was slower than a serial implementation. I managed to make an implementation that was slightly faster
than serial, but only when using a much longer list of numbers than the one specified, and only large numbers.
Meanwhile the multiprocessing implementation gave everyone speedups quite quickly.