
Simple and interpretable supervised machine learning of scRNA-seq cell types

The scRNA-seq field has reached a second wave, were the first initial systems under investigation are getting repeated. Either to ask more specific questions, or to get better data with the newer technologies available. This is highlighted in particular in a recent paper by Kiselev & Hemberg. They point out that we need to start thinking about cell type references similar to how there are genome references, and we need a way to map data to this reference.
I was wondering how a stereotypical machine learning multi-class classification model would perform for this task. Since the online scmap tool from the K&H paper comes with a couple of well annotated example data sets of pancreatic cells, this ended up being quite straightforward.
What we will do is train a machine learning model to predict cell types using one of the data sets, and predict cell types of cells from the other dataset with it.
The most basic multi-class classification model is Logistic regression, and we will use the implementation in scikit-learn. The entire analysis is in a notebook on Github, but let's walk through the key parts here.
To train the model, we will use the data from Segerstolpe et al, consisting of 3,500 cells annotated with 15 cell types. We want to predict the cell types of the samples using the gene expression values. First we split up the data so we can evaluate the model afterwards.
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = \
train_test_split(s_exprs, s_sample_info['cell_type1'], test_size=.2)Next we initiate the model.
from sklearn.linear_model import LogisticRegression
lr = LogisticRegression(C=0.05, penalty='l1', n_jobs=-1)First of all, we use L1 penalty in the model. This means we are favoring sparsity. That is, we believe only a small number of the genes determine the cell types, and we favor many genes having 0 weights. The C parameter determines how strongly we enforce sparsity. I picked 0.05 after trying a couple of different values.
Next we train and investigate the model, this takes about 5 seconds.
lr.fit(X_train, y_train)
LogisticRegression(C=0.05, class_weight=None, dual=False, fit_intercept=True,
intercept_scaling=1, max_iter=100, multi_class='ovr', n_jobs=-1,
penalty='l1', random_state=None, solver='liblinear', tol=0.0001,
verbose=0, warm_start=False)
lr.classes_
array(['MHC class II', 'PSC', 'acinar', 'alpha', 'beta', 'co-expression',
'delta', 'ductal', 'endothelial', 'epsilon', 'gamma', 'mast',
'not applicable', 'unclassified', 'unclassified endocrine'], dtype=object)
lr.coef_.shape
(15, 23171)The cell types we want to be able to predict gets stored in the lr.classes_ field. Logistic regression works by predicting a probability of a sample coming from a given class. In the standard version in sklearn, this is done by making oen binary logistic regression for each class, where logistic regression depends on a linear combination of weights times gene expression values. The class with highest probability gets assigned as the predicted class when evaluating the model on a new observation. The weights for each gene for each cell type is stored in lr.coeff_.
First let's have a look at the performance of the model
lr.score(X_train, y_train)
0.98256848096762717
lr.score(X_test, y_test)
0.92887624466571839I think this is pretty good. For the data used for training, the model is 98% accurate, while it is 92% accurate for the held out testing data. It should be noted that this might not be the best metric here, because the cell types are very different in number of representatives.
To predict using our model, we just use the lr.predict method.
y_hat = lr.predict(X_train)
y_hat
array(['ductal', 'alpha', 'not applicable', ..., 'not applicable', 'beta',
'not applicable'], dtype=object)The most straightforward way to investigate how the model is doing is by making a matrix of how different cell types get predicted.
from sklearn import metrics
pd.DataFrame.from_records(metrics.confusion_matrix(y_train, y_hat),
index=lr.classes_)
In particular we notice that some of the not applicable and unclassified cells get predicted as other cell types.
A particularly nice thing with linear model such as logistic regression is how interpretable they are. The weights of the genes directly relate to how the the cell types are predicted. Let's assign each gene as a marker for the cell type it's the strongest predictor of.
marker_genes = pd.DataFrame({
'cell_type': lr.classes_[lr.coef_.argmax(0)],
'gene': X_train.columns,
'weight': lr.coef_.max(0)
})
marker_genes.query('weight > 0.').shape
(628, 3)The final row tells us that of the ~23,000 genes we used as input, only 628 are used in predicting the cell types. Let's print out the top predictive genes for each cell type.
top_markers = \
marker_genes \
.query('weight > 0.') \
.sort_values('weight', ascending=False) \
.groupby('cell_type') \
.head(6) \
.sort_values(['cell_type', 'weight'], ascending=[True, False])
figsize(10, 20)
for i, m in enumerate(top_markers.cell_type.unique()):
plt.subplot(10, 3, i + 1)
g = top_markers.query('cell_type == @m')
plt.title(m, size=12, weight='bold')
for j, gn in enumerate(g.gene):
plt.annotate(gn, (0, 0.2 * j), )
plt.axis('off')
plt.ylim(6 * 0.2, -0.2)
plt.tight_layout()
We wrote before that logistic regression predicts the probability of each cell type. This can also be used as a visualization. After sorting the cells according to the known cell type, we can predict the probability, then plot the probability of each cell type for each cell.
shift_idx = y_train.argsort()
sorted_idx = y_train.sort_values().index
y_prob = lr.predict_log_proba(X_train.loc[sorted_idx])
Now let's finally get to the task at hand: treat this model as a reference, and predict cells from another dataset. The second dataset is from Muraro et al. This is 2,100 cells annotated with 10 cell types, the interesting point is to see if these cell types gets predicted in a reasonable way by our model.
Something we need to make sure of is that the genes in the new dataset are in the same order as in the previous. If a gene is not present in the new dataset, we set those values to 0.
X_new = m_exprs.T.loc[X_train.columns].T.fillna(0)
m_sample_info['predicted_cell_type'] = lr.predict(X_new)
m_sample_info \
.groupby(['cell_type1', 'pred_cell_type']) \
.count().iloc[:, [0]] \
.unstack().T \
.fillna(0)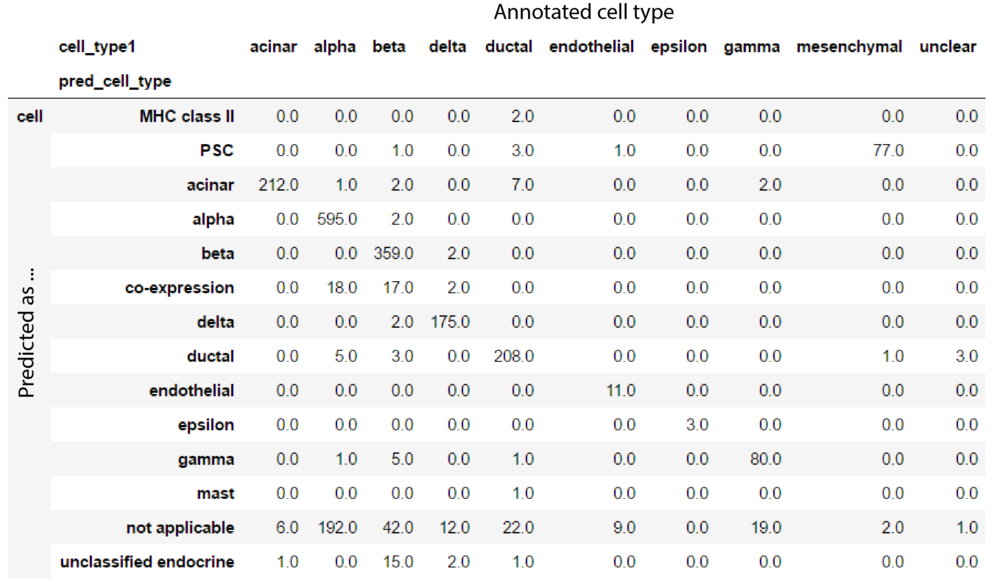
This is pretty nice I think! We didn't do any normalisation or batch correction etcetera, but the results still seems consistant. Based on this I think it's pretty easy to envision servers with models for cell types based on huge amounts of data that can be used by researchers to query new samples against.
I think clustering and cell type annotation will be considered similarly to transcriptome assembly and annotation in the future. An application which is certainly feasible, but a level more advanced than most users will need.
Again, this sort of analysis is pretty straight forward, and the notebook is available here
