
Low mapping rate 1 - Unsorted FASTQ pairs

Occasionally when working with scRNA-seq data, you notice that there is a large degree of heterogeneity in terms of the percent of mapped reads per cell. Typically this is one of the criterions for exculding cells from analysis. Usually we are pushed to get things done, and as long as we have enough cells with enough mapped reads to perform proper analysis we let it be and move on.
In our review of the history of scRNA-seq experiments we point out that the sequencing itself is one of the main current bottlenecks for large scale experiments. In light of this, I thought it would be useful to actually note what is causing us to sequence reads which we are not using in the actual analysis. I will write a series of posts with a number of contributing factors for low mapping rates I have noticed recently.
Unsorted FASTQ pairs
Typically we quantify gene expression with Salmon, and have some simple tools to extract QC data from the result files. One good combination of variables to look at in your cells is the number of mapped reads compared to the % of mapped reads.
In a recent case, we had a plot like this

What sticks out here is the large gap between high-mapping and low-mapping cells. It usually shows more of a continuous trend, with a more clear cluster of "proper" cells.
It turned out that the at some point in the data processing steps prior to quantificiation, the order in the reverse and forward FASTQ files had not been kept consistent. Example:
$ head -n 8 JE1704_C27_R{1,2}.fastq
==> JE1704_C27_R1.fastq <==
@NS500239:235:HL7JLAFXX:1:11101:21276:1083 1:N:0:ATGCGCAG+NTTAATAG
CCTCTACCCAGAGGCCCAGTGGCAGAGGCCTGGACAAGTATTGAACACAAGAACTGTAGTGGTCAGAGGGACTTAA
+
AAAAAAEEEEEEEEEEEEEEEEEEEEEEAEEEE/EEEEEEEEEEEEEEEEEAEEEEEEEEEEEEEEEEEEAEAEEA
@NS500239:235:HL7JLAFXX:1:11101:9041:1085 1:N:0:ATGCGCAG+NTTAATAG
TGTTTTTATTGATTTAGTCTGTTTCAGAGTCAAGGTGTCAACGAGGAAGGATGGATATCCATGGAGGAAGAAGAGA
+
AAAAAEE6EEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEEEEEEEEAEEAEEEEEEEAEEE/EEA/EA<E
==> JE1704_C27_R2.fastq <==
@NS500239:235:HL7JLAFXX:2:11101:17928:1074 2:N:0:ATGCGCAG+NTTAATAG
ACCTAATATAGCAGGTGGCCAGGACTGGGATCCAGCTGCCTGGATCAGGTCAGGCTTGAGGAAGACTGCTTAAGAG
+
AAAAAEEEEEEEEEEEEEEEEEAEEEEEEEEEEEEEAEEEEEEEEEEEEEEEAEEEEAEEEEEEAEEAEEEAE6/A
@NS500239:235:HL7JLAFXX:2:11101:5780:1075 2:N:0:ATGCGCAG+NTTAATAG
TATCAACGCAGAGTACGGGATGAGGTGTGGGACGCACTGCCAGCGTTGCCCTTTAGTATAGCTCTTGGTAAACTAA
+
AAAAAEAEEEEA6AEE//EEEA///EE//E/EEAEEA6A6EEEEE//6E/<EAE<EEE6E/EEEAE/EEEEEEEE6If everything is correct, the headers of the FASTQ records should be identical up to the first space. The number after the first space indicated if the FASTQ is a forward read or reverse read. Here we specifically see that the reads come from different sequencing lanes (the number after HL7JLAFXX:).
When Salmon is mapping with umatched forward and reverse reads, the majority of these will map to different transcripts from eachother. This will cause the read to be considered unmapped as it is an event which is not consistent with typical RNA-seq libraries.
The solution to this problem is pretty simple: just sort all your FASTQ files by the header header. The quickest solution I stumbled upon for this is from the EdwardsLab blog which suggests a Bash oneline to do this.
After sorting, the beginning of the FASTQ pair above looks like this
$ head -n 8 JE1704_C27_R{1,2}_sorted.fastq
==> JE1704_C27_R1_sorted.fastq <==
@NS500239:235:HL7JLAFXX:1:11101:10008:20335 1:N:0:ATGCGCAG+CTTAATAG
CCCACAGCCTCTGCCGCGGGTACCATGAAGATCTCTGCAGCTGCCCTCACCATCATCCTCACTGCAGCCGCCCT
+
AAAAAEEAEEEEEEEEEEEEEE//EEEEEEEEEEEEEEAEEEAEEEEAA66EEE/E/EEEEEEEEEEEEAAEEE
@NS500239:235:HL7JLAFXX:1:11101:10009:10878 1:N:0:ATGCGCAG+CTTAATAG
TACCAACACATGATCTAGGAGGCTGCTGACCTCCAACAGGAATTTCACCACTTAACCCTCTAGAAGTCCCACTA
+
AAAAAEEEEEEEEEAEEEEEEEEEEEEEEEEEEEEEAEEEEAEEEEAEEEEEEEAEEEEEE/EEEEEEEEAEEE
==> JE1704_C27_R2_sorted.fastq <==
@NS500239:235:HL7JLAFXX:1:11101:10008:20335 2:N:0:ATGCGCAG+CTTAATAG
TGGTATCAACGCAGAGTACGGGGGTGCAGAGGGCGGCTGCAGTGAGGATGATGGTGAGGGCAGCTGCAGAGATCTT
+
A/AAAEEEAEEEEEEEEEEEE/EEEEEEEEEEEEEEEEEEEEEAEEEEEEEEEEEEEAEEEAAAEEEEEAEEEEEE
@NS500239:235:HL7JLAFXX:1:11101:10009:10878 2:N:0:ATGCGCAG+CTTAATAG
ATTACATGGAGTCCATGGAATCCAGTAGCCATGAAGAATGTAGAACCATAGATACCATCTGAAATGGAGAATGATG
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEEE/EEEAAEEEEEEAAEEEEEEEEEEEWhat caused the files to not be correctly sorted to begin with? it could have been something about how the files from different sequencing lanes were merged. I have also had issues with this happening befoer when using the samtools fastq command on CRAM files which have been sorted by alignment coordinate.
It seems a small number of the cells had succesfully kept the FASTQ order, explaining the "outlier" population at 80%.
After sorting all files and rerunning Salmon, we get much more reasonable mapping rates.

There are still some cells with lower mapping rates, but not nearly as many as before. And we see a more consistant cluster of highly mapping cells at ~80%.
We can also visualize how much the mapping improved for each cell when properly ordering the FASTQ files.
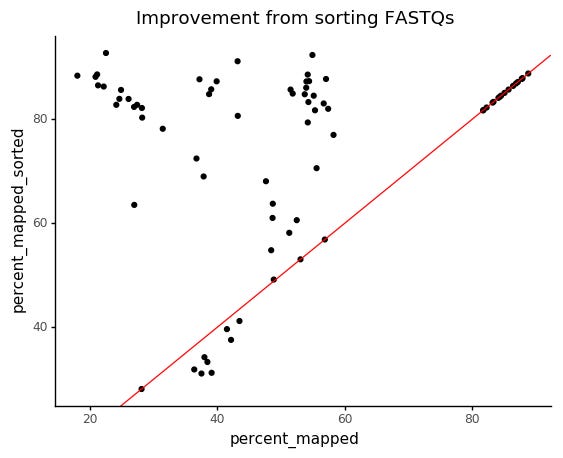
This was probably the cause of low mappting rate which is easiest to deal with, and it was introduced at the data processing side. But a good thing to keep in mind, if you have consistently very low mapping rate, have a quick look to see that reads are sorted correctly.
