
Count based autoencoders and the future for scRNA-seq analysis

Two recent computational methods, scVI by Lopez et al and DCA by Eraslan & Simon et al, are the most promising methods for large scale scRNA-seq analysis in a while. This post will describe what they do and why it is exciting, and demonstrate the results of running them on three recent datasets.
In almost all cases, to do anything useful in scRNA-seq the tens of thousands of genes measured need to be summarised and simplified. An extremely effective way to summarise many variables through their covariance structure is principal component analysis (PCA). However, scRNA-seq data consists of counts, which have particular behaviours that cause issues with the interpretation and of PCA.
Potential ways of dealing with this is either figuring out how to transform count data to emulate the characteristics of continuous Gaussian data, or to reformulate PCA for the count setting. While data transformations have historically had some success, they don’t perform so well for low count numbers, which is the case when massive scRNA-seq experiments are economically feasible.
A couple of years ago Risso et al successfully created a generalized linear factor analysis model for scRNA-seq counts called ZINB-WaVE, based on a zero-inflated negative binomial (ZINB) count distribution. Omitting some further features, in this model underlying rates of observations of mRNA molecules from specific genes are modelled by a low-dimensional collection of continuous factors. Every cell has a particular hidden value for these factors, and to fit the model all cells are investigated and assigned the most likely factor values. (In these equations, red color indicates parameters that need to be inferred.)
With these ZINB based methods, the data does not need to be scaled, nor normalised, and in principle the common step of selecting highly variable genes is not necessary. The data just plugs in.
Inherently, learning the X means that each cell is compared to all other cells. The factors (W) can be investigated in attempts to deduce meaning, and in that way we gain knowledge. But if you show the model a new cell y, it doesn’t know what to do with it. The inference will need to be rerun with the entire dataset including this new cell.
Two new methods, called scVI (single cell variational inference) and DCA (deep count autoencoder) rethinks this model, by moving from factor analysis to an autoencoder framework using the same ZINB count distribution. (Their titles and abstracts phrase them as imputation methods, which is a bit odd and substantially undersell them!) The two methods have slightly different parameterizations, but conceptually (abusing notation a bit), this represents with they both do:
A parametric function from the observed space (gene counts) to a low-dimensional space is fitted (g), at the same time as a function that maps the low dimensional space to ZINB parameters (f). For any given cell, you can apply g to get its X-representation, then apply f on that to get parameters, and evaluate the likelihood of the cell. The functions g and f are parameterised as neural networks because these are flexible and efficient.
This unlocks a lot of benefits. For inference, this setup makes it easier to use stochastic optimization, and is directly compatible with inference on mini-batches: you only need to look at a few cells at a time, so no matter how many cells you have, you will not run out of memory. Scientifically, this allows you to generalize. You can apply g to any new cell and see where it ends up in the X-space.
By analysing how different regions of the X-space map to gene expression, markers and differential expression can be investigated. And on the converse, if you perform clustering in the X-space, you can take new cells and evaluate which clusters the g function maps them to.
To illustrate what the methods do, we will apply them to three recent datasets. I picked smaller datasets on the order of ~2,500 cells because I wanted to quickly run them on desktop just for testing. One from Rosenberg et al 2018, where I randomly sampled 3,000 out of 150,000 developing mouse brain cells. The second one is a taxon of Peripheral sensory neurons from mousebrain.org, which is part of Zeisel et al 2018. And finally a dataset of male mouse germ cells that I found on GEO, but I couldn’t find an associated paper for (Lukassen 2018). I ran both DCA and scVI with default parameters: DCA produces a 32-dimensional representation, and scVI a 10-dimensional. To qualitatively inspect the results I ran tSNE on the representations, and colored the cells based on labels provided from the data sources.
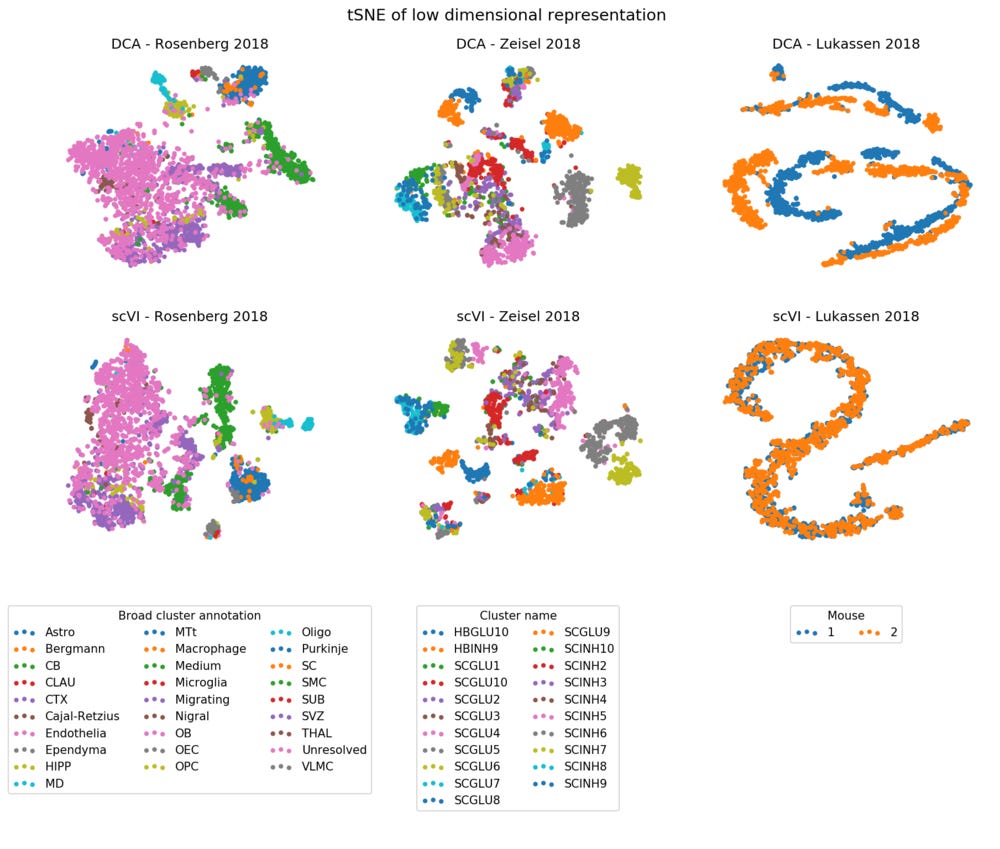
The DCA method is implemented around the anndata Python package, and is very easy to run on any data you have. The scVI implementation requires you to manually wrangle your data into TensorFlow tensors with correct data types, which can be frustrating if you are not used to it. This does however imply that if you want to scale the inference using out-of-core strategies, scVI directly supports that.
In terms of run time, DCA was much faster than scVI, finishing in a few minutes. scVI took about an hour for each dataset. A large component is probably that DCA implements automatic early stopping, while scVI will run for as many epochs as you tell it, even if the fit doesn’t improve.
The scVI method has the option to account for discrete nuisance variables (batch effects), but I did not try this. And even without it, it seems to align the two mice quite well in the Lukassen 2018 data!
I am curious if there is a way to also account for continuous nuisance parameters (e.g. amplification cycles). In ZINB-WaVE this is straightforward, because it is a GLM, but it is not so clear here. I mentioned clustering in the X-space, it might be possible to formulate these models as structured autoencoders (SVAE), and encourage the g function to learn a representation that favours cell type segmentation. Notebooks where I ran the methods on the different datasets are available here.
