
Handling confounded samples for differential expression in scRNA-seq experiments

Typical single cell RNA-seq studies are still working in an exploratory sense, aiming to characterize the diversity in a cell population by identifying discrete cell types or trajectories of variation. But there are also efforts trying to identify how different experimental or observed conditions affect different cell types. For example, by sorting blood cells from healthy and diseased people, scRNA-seq could in principle be used to assign cell types to the cells, and differential expression for each cell type could be investigated to see if the disease is affecting any particular cell type more than others.
The task of differential expression in essence boils down to creating a model of the form \( \text{expression} = b \cdot \text{[is diseased]} + a \) and investigating whether the \( \text{[is diseased]} \) label helps predicting the gene expression level.
In such a procedure, each individual contributes many cells of each cell type. So in total, the number of observations (cells) might be in the thousands. The number of individuals contributing the cells might be much smaller, and this raises the question of how to think about statistical power for the test. It is important that the between-individual variation is not larger than the between-conditions variation.
When comparing two assigned cell types against each other it is straightforward to account for between-individual variation. Since (ideally) each individual contributes with samples of each cell type, an individual-specific effect can be added to the model. In other words it means writing a model of the style \( \text{expression} = b \cdot \text{[is cell type IV]} + \sum_{i} c_i \cdot \text{[is mouse i]} + a \).
If the goal is to compare gene expression between healthy and diseased mice for a particular cell type, it is no longer possible to separate the individual effect from the disease effect. A model would be \( \text{expression} = b \cdot \text{[is diseased]} + \sum_{i} c_i \cdot \text{[is mouse i]} + a \), but whether the disease effect goes into the \( b \) parameter or the \( c \) parameter is ambiguous. There is complete confounding between condition and individual. If you compare this model to \( \text{expression} = \sum_{i} c_i \cdot \text{[is mouse i]} + a \) you will not be able to tell a difference. We would for example never have data from the same individual being both healthy and diseased.

This means that when accounting for individual-specific effects on gene expression, it will never be possible to get low p-values. Not without making the assumption that the individuals are directly comparable, and we do not need to consider individual-level effects. This might be reasonable for lab mice, but when analyzing cells from human donors, there will be a lot of genetic and lifestyle variation between individuals.
A strategy to deal with this is use multilevel regression rather than linear regression. A simple way to think about these models is as means at different levels, with level-specific variance that observations vary around. Say there is an overall mean expression level, and a between-mouse variance. Then for each mouse there is a mean expression level, with between-cell variance for that particular mouse.
Such a model can be compared to a model where there is an overall mean, with condition-specific variance. Then each condition (healthy vs diseased) has a mean, as well as between-mouse variance for the conditions. And in turn, each mouse, within a condition, has a specific mean with between-cell variance. If grouping the mice by disease give a better model fit for the gene expression values, then this is evidence of the gene being perturbed by the disease state. While also accounting for between-individual variation!

To test this approach to differential expression, I looked at a dataset published by Hrvatin et al 2018 [1]. They perform scRNA-seq on cells from the visual cortex of mice. These are mice that have been living in complete darkness for several days. Some of the mice are exposed to light for an hour before they are sacrificed and cells are collected. The concept is that now you can identify which cell types in the visual cortex get activated by the exposure to light by investigating the differential expression of no light vs 1 hour light exposure.
In the paper they describe that the most dramatic effect was in the cells annotated as ExcL23, so I sorted these out, and only picked cells with either 0h or 1h light stimulation. In total this data has 1,856 cells from 17 mice, where 9 mice were not exposed to light and 8 mice were exposed to light for 1 hour.
I used three different models for testing differential expression. An ordinary linear regression model on log(counts + 1) expression values using the R lm function; a Poisson generalized linear model on observed counts using the R glm function; and a Poisson multilevel model on the observed counts using the glmer function in the lme4 R package. In formula notation, these would be the models:
LM H0: ‘log(expr + 1) ~ log(total_count)’
LM H1: ‘log(expr + 1) ~ log(total_count) + stim’
GLM H0: ‘expr ~ offset(log(total_count))’
GLM H1: ‘expr ~ offset(log(total_count)) + stim’
LME H0: ‘expr ~ offset(log(total_count)) + (1 | sample)’
LME H1: ‘expr ~ offset(log(total_count)) + (1 | stim/sample)’To obtain p-values, I performed likelihood ratio tests between the H0 and H1 models. Note that the multilevel model is the only one aware of sample (mouse) grouping!
In the results I am highlighting genes that Hrvatin et al also point out as particularly differential between no light and 1h light.

Ordinary regression clearly identifies the genes discussed by Hrvatin et al. But the p-values are extremely small (for reference, the red dashed line indicate p=0.05). The model is not acknowledging the fact that cells from the same mouse probably have a lot of internal correlation, and therefore the model fit is overly certain. The same phenomenon can be observed in the GLM model. The multilevel model has much more reasonable p-values.
An alternative proposed strategy is a two-step one. First, define cell types on the single cell level from scRNA-seq. Then, for each cell type sum up the counts for each biological sample (mouse) and treat them as “pseudobulk”, as if they were sorted cells from each sample. Now we can apply differential expression tests on the summed level. I applied the same models, but for such summed pseudobulks, now having only 17 observations rather than 1,856.
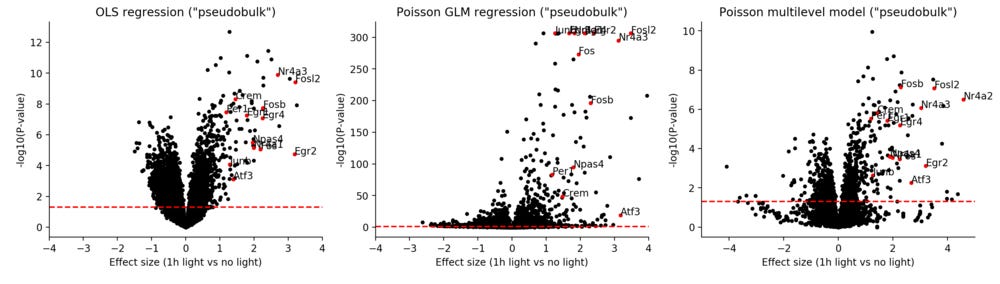
The OLS regression now seems much more reasonable, the GLM model still has the same issue of seeming overly confident, and interestingly, the Poisson multilevel model for the pseudobulk gives exactly the same results as for the single cell data. This idea of pseudobulk analysis was explored by Lun & Marioni 2017 [2].
There’s no particular ground truth here for a proper comparison, but the investigation suggests that it is possible to use multilevel models for differential expression. I like that the multilevel approach ties directly to the hierarchical nature of the experiment. Here I only used basic R functions rather than specialized DE packages. The specialized packages would do more clever things like empirical Bayes shrinkage across the genes, or use more sophisticated offsets than the total_count I use here. I just wanted to compare these strategies directly with naive implementation. (For an interested reader, I suggest the recent comparison by Soneson & Robinson 2018 [3].)
Finally, I used Poisson models here; there are plenty of reasons to think that negative binomial models would be more appropriate, and I might revisit these questions to test that at some point. Notebooks, R scripts, and other material for making this post are available on Github. Thanks to Lior Pachter for editorial feedback on this post.
References
[1] S. Hrvatin et al., “Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex,” Nat. Neurosci., vol. 21, no. 1, pp. 120–129, Jan. 2018.
[2] A. T. L. Lun and J. C. Marioni, “Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data,” Biostatistics, vol. 18, no. 3, pp. 451–464, Jul. 2017.
[3] C. Soneson and M. D. Robinson, “Bias, robustness and scalability in single-cell differential expression analysis,” Nat. Methods, Feb. 2018.
