
Gene Discovery Rate in RNA-seq data

I was curious regarding at what rate new genes are detected as the number of reads in RNA-seq data increases. Let’s start by looking at the results.
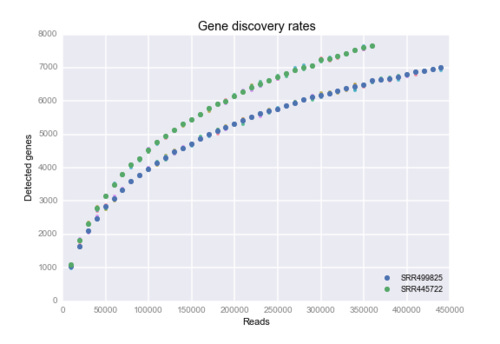
The figure shows how many genes there are with nonzero counts at certain read depths.
In particular I was interested how this discovery rate looked for single-cell RNA-seq data. I found some read files at ENA which was from human cells. The two data sets used are SRR445722 and SRR499825.
In the interest of saving space on my tiny laptop, I only kept the first four million reads in the fastq files. For each read number investigated, reads are first randomly sampled from the fastq file using the reservoir sampling of seqtk. The sampled reads are then mapped to the human transcriptome (Homo_sapiens.GRCh37.73.cdna.abinitio.fa) using bwa mem. To map from transcripts to genes, I use eXpress (with the annotation file Homo_sapiens.GRCh37.73.gtf.gz). The final step is to count the number of genes which have counts different from 0.
In the interest of time and simplicity, I didn’t perform any quality or adapter trimming of the data.
For each read count, the sampling was performed five times with different random seeds. In the figure these replicates can barely be seen, but at some pints some smaller colored points can be glanced.
These are the pipeline for the process for the two datasets:
for READS in $(seq 10000 10000 1000000) do for i in $(seq 5) do echo $READS reads >> sample_num_genes_SRR445722.out seqtk sample -s$(date +%s) SRR445722_4M_reads.fastq $(echo "scale=8; $READS / 4000000" | bc) | \ bwa mem -t 2 Homo_sapiens.GRCh37.73.cdna.abinitio.fa - | \ express --no-bias-correct Homo_sapiens.GRCh37.73.cdna.abinitio.fa cut -f5 results.xprs | grep -v "\b0\b" | wc -l >> sample_num_genes.out done done
and
for READS in $(seq 10000 10000 1000000) do for i in $(seq 5) do echo $READS reads >> sample_num_genes_SRR499825.out SEED=$(date +%s) FRAC=$(echo "scale=8; $READS / 4000000" | bc) bwa mem -t 2 Homo_sapiens.GRCh37.73.cdna.abinitio.fa \ <(seqtk sample -s$SEED SRR499825_1_4M_reads.fastq $FRAC) \ <(seqtk sample -s$SEED SRR499825_2_4M_reads.fastq $FRAC) \ | express --no-bias-correct Homo_sapiens.GRCh37.73.cdna.abinitio.fa cut -f5 results.xprs | grep -v "\b0\b" | wc -l >> sample_num_genes_SRR499825.out done done
They are slightly different since SRR499825 is paired-ended while SRR445722 is single ended.
I ran these for as long as I could leave my laptop on and stationary at a time.
What I find interesting about this is how these logarithm-like curves seem are so smooth.
Both studies these data come from have plenty of replicates, so a more thorough version of this study would be to try data from multiple cells and make sure that correlates. It would also be interesting to see how bulk RNA- seq compares to these data sets. I also only looked at data from human cells because I didn’t want include another set of transcriptome and annotation file to keep track of, but there are a lot more mouse single-cell RNA-seq studies on ENA than human.
It would also be fun to run the analysis much longer, to get up to millions of reads, where we start seeing the majority of genes as detected. And see precisely at which point this starts to happen.
EDIT

I reran the computations, but this time I let both data sets run to completion (depth of 1000 000 reads). I also saved the counts for each gene, so I could query detected genes with a lower cutoff. The new graph above show the detection rates for lower cutoffs of one, three and five mapped reads.