
Improving SCVI for low-count cells through self-supervised augmentation

To improve utility of cells with low UMI counts, I wanted to optimize KL weighting in SCVI models. It turned out that the issues with analyzing lower-UMI cells with SCVI is not due to posterior collapse from the KL term, but instead reflects a learned behavior of the encoder in the SCVI model. By including a joint embedding cross correlation loss objective, the encoders achieve better performance for low-UMI cells. All the details are in the resulting paper, available at https://www.biorxiv.org/content/10.64898/2026.02.11.705441v1. This post summarizes the main results.
In an earlier post I demonstrated that when a cell has low UMI counts, an SCVI model will place them in the center of the representation space. In that post, I used a small subset of genes and looked at how representations in a 2-dimensional SCVI model depended on UMI depth.
In that post, the low-count behavior could be explained by the balance between the negative binomial reconstruction likelihood and the KL divergence with the prior, which pulls representations to the center. When counts were low the KL term dominated.
Using the small set of 141 genes, you could see which total UMI counts you needed to beat the KL term and move cells away from the prior in the center of representation space. As a follow-up, I wanted to understand what kind of UMI library sizes you need to beat the prior in actual transcriptome wide data that I’d use for analysis. This way I could see what lower limits of useful total UMI counts are, and maybe I could put a lower weight on the KL term in the SCVI model to make it work better for cells with lower counts. This would enable analysis with cheaper and lower quality data.
It turns out, the KL term is not responsible for the low-UMI clustering behavior! In fact, low-UMI cells don’t converge towards the center. Instead, the SCVI encoder defines a bias point f([0, ..., 0]), where a hypothetical cell with zero counts from all genes is placed. This bias point is implicitly learned by the SCVI encoder during training, and it can be as far away from the center of representations space as high-UMI cells.
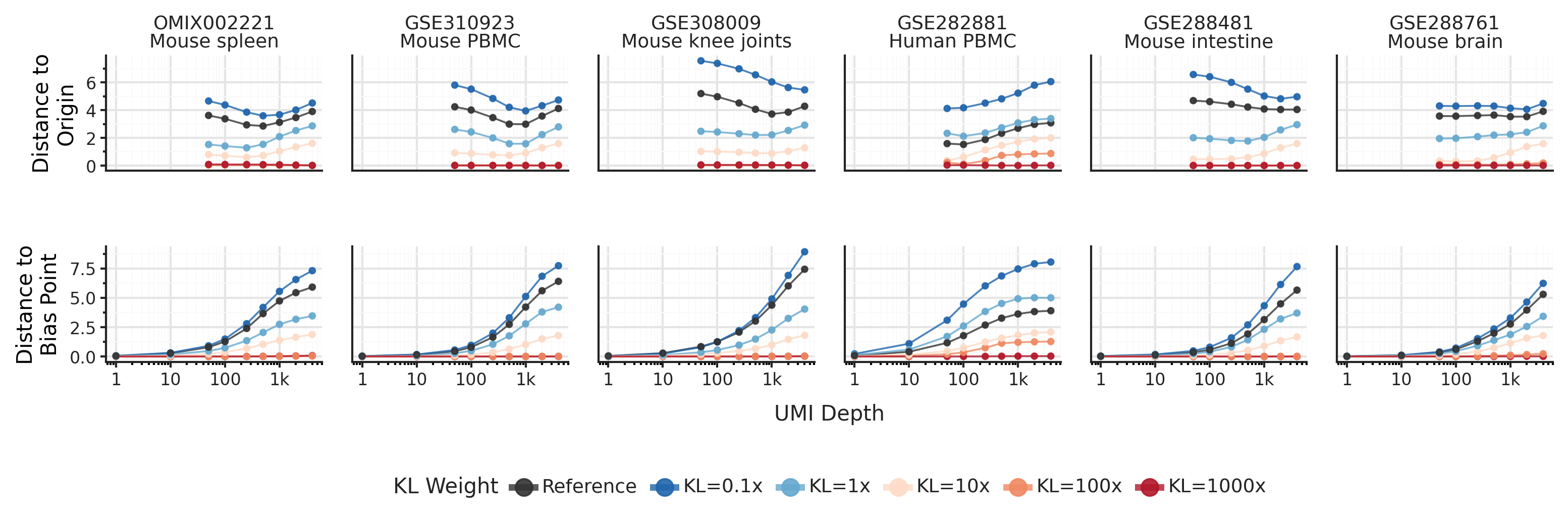
It turns out the default KL term is far too weak to assert meaningful influence on the representations. To force convergence towards the center of representations space it needs to be upweighted about 100X. At that point, you will also start observing posterior collapse where the model fails to learn meaningful representations.
Lowering the KL weight on the other hand, does _not_ reduce convergence towards the bias point as total UMIs per cell decreases. So if we want to improve SCVI for cells with lower UMI counts, what can we do instead?
The way I investigated the behavior was by subsampling UMI counts of cells using binomial thinning. One idea was to include this thinning procedure during training, increasing the depth-diversity the model sees. This didn’t improve the the encoder: it still showed similar or worse performance on downstream tasks.
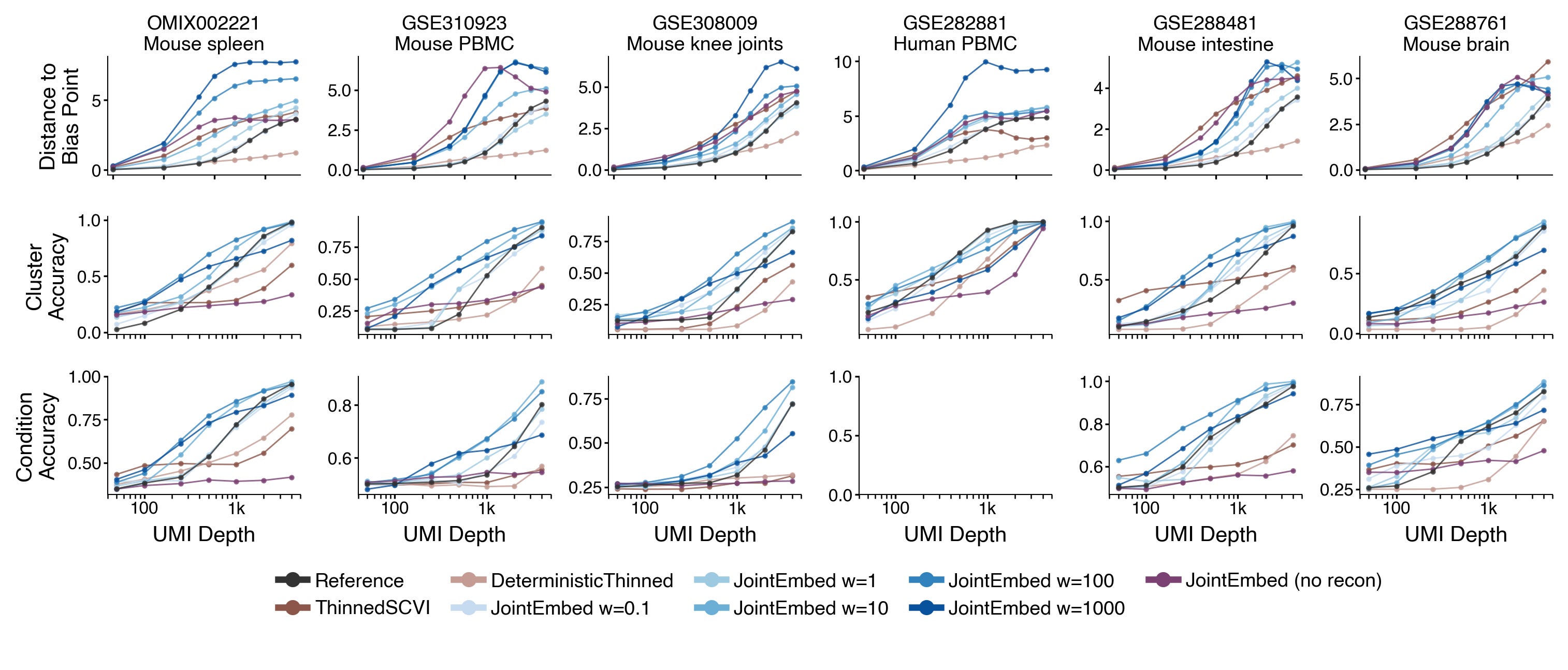
Instead of just showing the model more diverse training data, I added a joint embedding loss. By passing original counts y and thinned counts y* through the encoder to make representations z and z*, we can then use a loss that penalizes the model when it embeds z and z* far apart from each other. In particular I used a cross correlation loss to simultaneously encourage close representations of z and z* and avoid posterior collapse.
This strategy makes the model learn encoders that slows down the convergence towards the bias point as UMIs per cell decreases. This directly leads to improved performance in terms of preserving cell type identity or experimental conditions in the learned representations.
Looking at reconstruction performance, the inclusion of the joint embedding loss has minimal impact on the ability to normalize gene expression or simulate gene UMI counts.
With this additional loss, QC thresholds for data used in analysis can be lowered, and data from precious archival samples can be used more effectively.
I also investigated if this joint embedding loss is sufficient. Maybe we don’t need to perform reconstruction at all to learn useful biological cell representations?
It turns out including the reconstruction loss is crucial. Models trained with joint embedding loss alone fails completely on downstream tasks like classifying clusters or separating experimental conditions.
The full paper is available at https://www.biorxiv.org/content/10.64898/2026.02.11.705441v1. The models are implemented in the SCVI branch at https://github.com/vals/scVI/tree/scvi-joint-embedding, and code for analysis and figures are available at https://github.com/vals/scvi-joint-embedding-reproducibility.
From around the web
