
Low mapping rate 2 - Ribosomal RNA

In the first post of the low mapping rate series I started off by describing a problem at the data processing level in a dataset. In the coming few posts I will focus on a particular dataset and iteratively increase the mapping rate due to different factors.
The data is from our study of CD4+ immune response to Malaria infection. In the study we first performed the experiment using the Fluidigm C1 system with the SMARTer kit, then we replicated the results using Smart-seq2 in microwell plates. Here I will use the Smart-seq2 data.
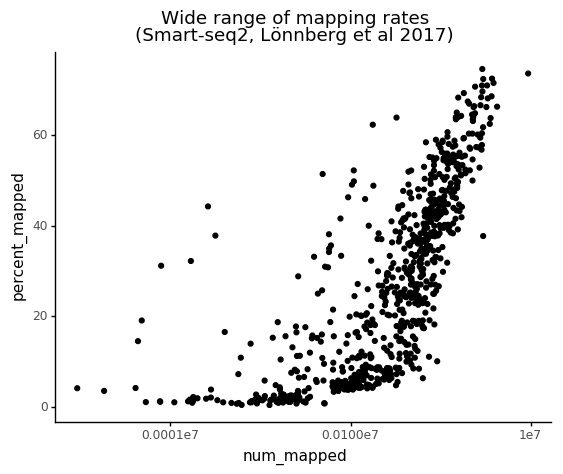
This data have a particularly large range of mapping rates for the individual cells, evenly distributed between 1% and 70%.
Ribosomal RNA
Ribosomal RNAs are highly abundant in cells, though unlike mRNA these are not polyadenylated. Since (almost) all scRNA-seq protols make use of oligo-dT sequences to reverse transcribe RNA to cDNA this is not a big issue.
The RNA component consists of a number of rRNA genes, repeated in chunks in various locations of the genome. These genes are 5S, 5.8S, 28S (all parts of the large subunit), and 18S (small subunit). In particular, 18S have a couple of (relatively short) stretches of poly-A in its sequence. My theory is that when the amount of mRNA is very limited in a sample the olig-dT binds these small stretches and the 18S gets reverse transcribed.
To investigate this, I added the sequences of Rn5S, Rn5.8S, Rn18S, and Rn28S from mouse together with the GENCODE transcripts and ERCC spike-in sequences in a new reference, and reran all the samples through Salmon. On average this had the effect of increasing the mapping rate, with a number of samples having almost twice the mapping rate as before.

The data here consists of cells from many individual mice, from different time points in the infection with a couple of replicates. By necessity of the technology cells from each mouse and time point need to be sorted into individual microwell plates. From the quantified gene expression/abundance values we can compare the controbuting sources in each individual cell, stratified by plate to see if there are any trends.

Here ENSMUS corresponds to contribution from the mouse transcriptome. The different rRNA genes are indicated. Here we see that Rn18S is contributing far more than the other genes. It is also clear that different samples (plates) have different contributions of rRNA.
If you haven't included ribosomal RNA in you mapping reference and are working on mouse, a red flag for rRNA contamination is particularly high expression of genes called CT010467.1, AY036118, Erf1, or Gm42418. These genes overlap a region on Chromosome 17 which have particularly similar sequnce to 18S. I have seen many datasets where any of these genes are the top 3-4 most highly expressed genes in a cell.
