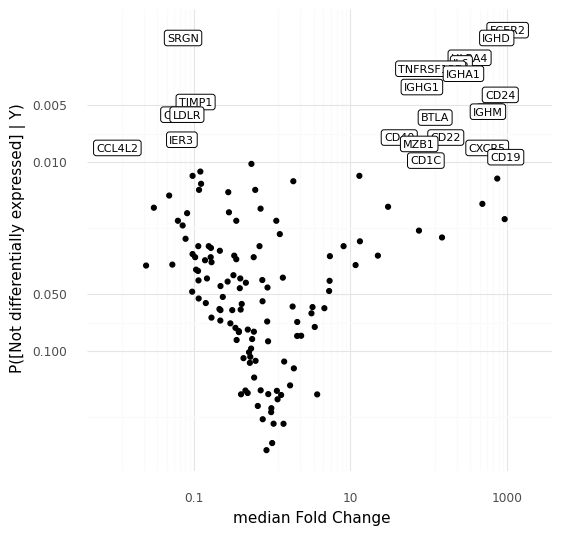
Tracking grocery expenses
An interesting trend is the increase in online services providing APIs with a cost per query. This seems particularly useful for data extraction on parsing tasks. Obtaining usable data from various sources is not necessarily difficult, but automated solutions such as data munging scripts can be tedious. Once you have finished a parser you tend to be sapped of the creativity you went in with for uses of the data.
I came across the parsing service Parseur, which charges a few cents to extract data from a document, in particular emails.
To test the service, I thought of something I have been curious about for a while: whether my monthly grocery spending has changed over the last couple of years. I order groceries from Whole Foods, and some time in the last year I changed the frequency of how often I get groceries. Since I don’t order groceries at regular intervals, and the intervals have changed over time, it is hard to compare order-to-order regarding how much I’m spending on them.
In my email inbox I had 106 Whole Foods order receipts starting from November 2019. I signed up for Parseur, and forwarded all of them to my Parseur email for the task. Over the years, the format of the receipt emails has changed slightly, and getting general parsing templates took some trial and error.
After parsing the emails, I could download a CSV file with the extracted information. One column in the CSV has the timestamp of the order, and another column has the order amount.
To account for variation in order frequency over time, created a running window of 91 days, and calculated the sum of the orders in each running window. By dividing the results by three, I get an average monthly grocery spend value.
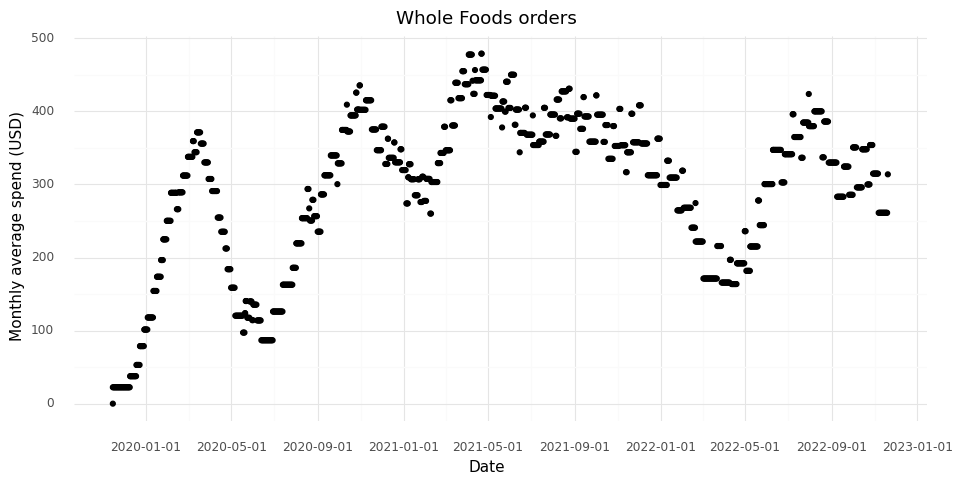
This way I learned that at the moment I am spending about $300 per month on groceries, with about +/- $50 month-to-month variation.
The first rise in the trend in 2019 is from starting to use the service and the running sum building up. The first drop is from the service breaking down at the beginning of the COVID pandemic, at which point limited deliveries were available. I don’t remember what happened at the end of 2020, but that was probably also related to the pandemic. Since then my spending was largely stable after the first quarter of 2021. The dip in the first quarter of 2022 is probably a reflection of a long vacation I had in April where I went to restaurants much more often than I usually do.
It took me a couple of hours getting the email parsing templates working. I also messed up in how I forwarded a number of the emails, making me waste ‘documents’ that I paid for parsing. In the end, I think I saved a lot of times compared to figuring out how to download the emails and parsing them by myself. I was pretty impressed by how well the GUI from Parseur worked for creating parsing templates.
The notebook for creating the plot is available on Github: https://github.com/vals/Blog/tree/master/221121-tracking-grocery