
SCVI with variational batch encoding

For last couple of months I have been exploring batch integration strategies with SCVI and MRVI, and the possibility to optionally disable integration when encoding single cells.
These models allow you to ask questions of the data you have trained the models one. But what if you have a pre-trained model and want to apply it to new data? You will not be able to integrate out new, unseen, batches. In this post I am exploring a strategy to solve this problem.
As a reminder, the conditional variational autoencoder used for batch integration in SCVI can be written as
In this formulation, z_n are the learned representations of the single cells, while s_n is an indicator for the batch identity. This allows the neural network f() to decode gene expression differently depending on the interaction between single cell representations and batch identities.
Practically, the default in SCVI is to represent a batch k as a one-hot encoding vector. The vector will have the length corresponding to the total number of batches K, and the element k being 1 in the vector indicates batch k (and all other entries are 0). Thus “one-hot”.
Of course, the vector s_n having length K means you can’t really do anything if you want to use the model with a new K+1’st batch from some new data. You are limited to the batches you trained the original model on.
There is an experimental option in the SCVI model called batch_representation with the two options 'one-hot' and 'embedding'. The default 'one-hot' option implements the behavior described above. The option 'embedding' implements a new behavior that learns a low-dimensional embedding for each batch. Practically, these embeddings are lookup tables that takes a batch indices and return the low-dimensional vector representing the batch. In this setting, s_i[n] are then these continuous vectors, whose values are parameters that are learned during training.
The 'embedding' option has an interesting feature: in theory, ‘similar’ batches should get proximal embedding vectors. This could be used to answer questions about batches. For example, if batches are patient samples, and patients have different diagnoses, you could learn which diagnoses are similar to each other.
These embeddings are finite-size lookup tables, and they are learned during training. We are still in the position that we can’t add new batches. It would be possible to extend look-up table with the new batch and run some training to learn the parameters for this batch, but this is impractical.
We can use this batch representation strategy as inspiration for a next step.
Remember that inference in the SCVI model is done through an inference model that takes as input observed data,
Here y_n is an array of observed UMI counts for cell n.
What if we can make a version of the SCVI model where batch embeddings s_n are encoded using another inference model?
This would give us variationally encoded batches. The known data X need to contain rich enough information for an inference model to be learned to encode the data to the variational batch representations.
A good candidate for the data X is the pseudobulk of all cells in batch i.
Let’s try this out!
Results
As in the previous posts, we will use the Asian Immune Diversity Atlas (AIDA, Tian et al 2024). The dataset has 1.1 million blood cells, but particularly important here, it has samples from 503 donors, making it realistic to learn an inference model even after holding out 50 donors as a test set.
After implementing the model with variational batch encoding, I split out all data from 50 donors as a test set. The remaining dataset was used to train a model with the default 'one-hot' option, a model with the experimental 'embedding' option, and finally a model with our new 'variational' option for batch_representation.
All models used 10-dimensional cell representations, the default for SCVI models. Both the 'embedding' and 'variational' batch representation models used 5-dimensional representations for donors. This is the default for the experimental 'embedding' option. All models were trained for 20 epochs.
In each training run the training data is further randomly split into training and validation. To make performance comparable, we evaluate the ELBO (evidence lower bound) for the full non-test fraction of the data. This will not let us evaluate overfitting, but it it does give us a global picture of average performance.
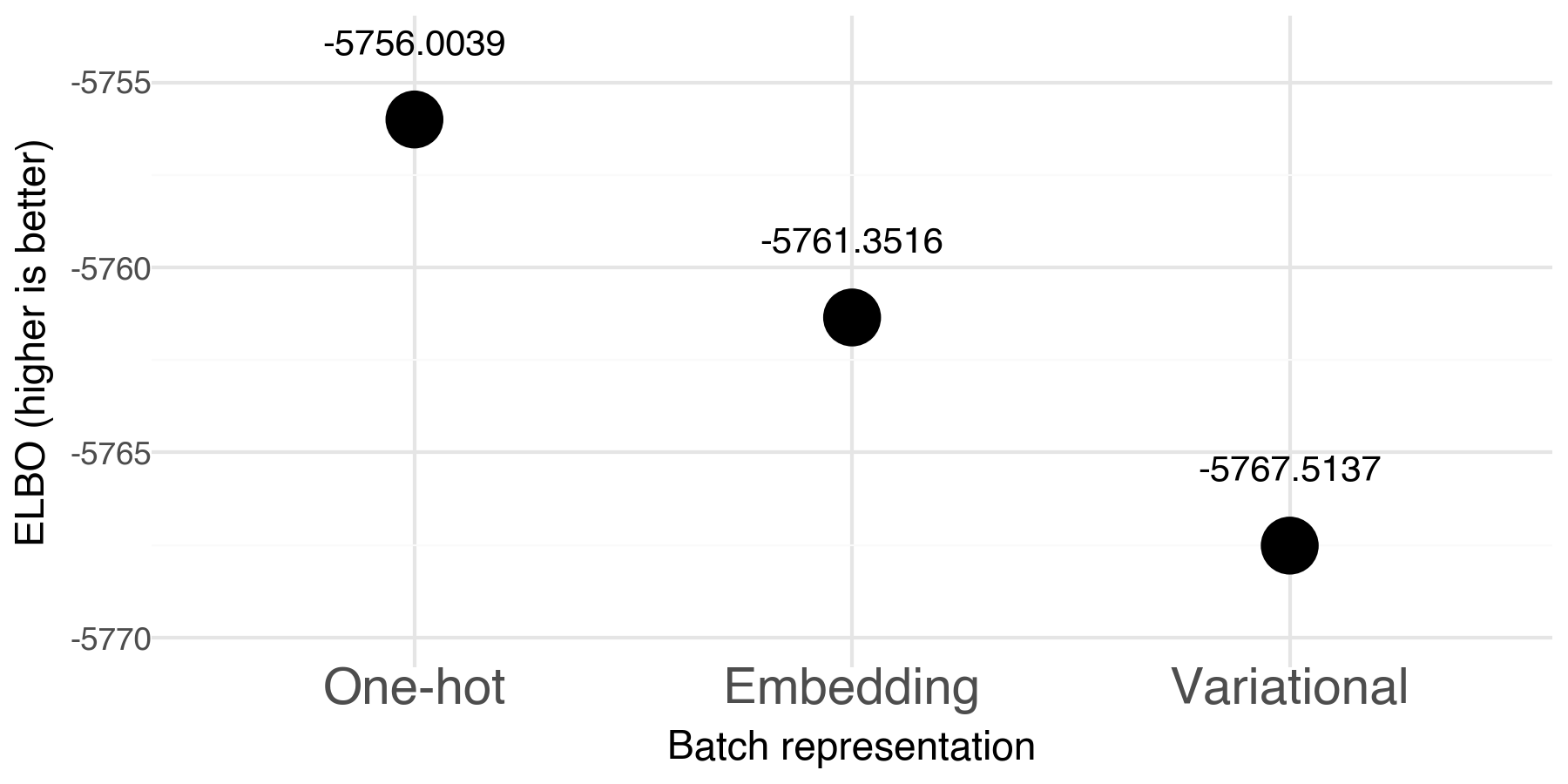
The default 'one-hot' option performs the best in terms of fitting the model to the data. The new 'variational' option performs the worst.
To get a feel for the batch integration performance of the different options, we can quickly visually inspect the cell embeddings using TSNE.
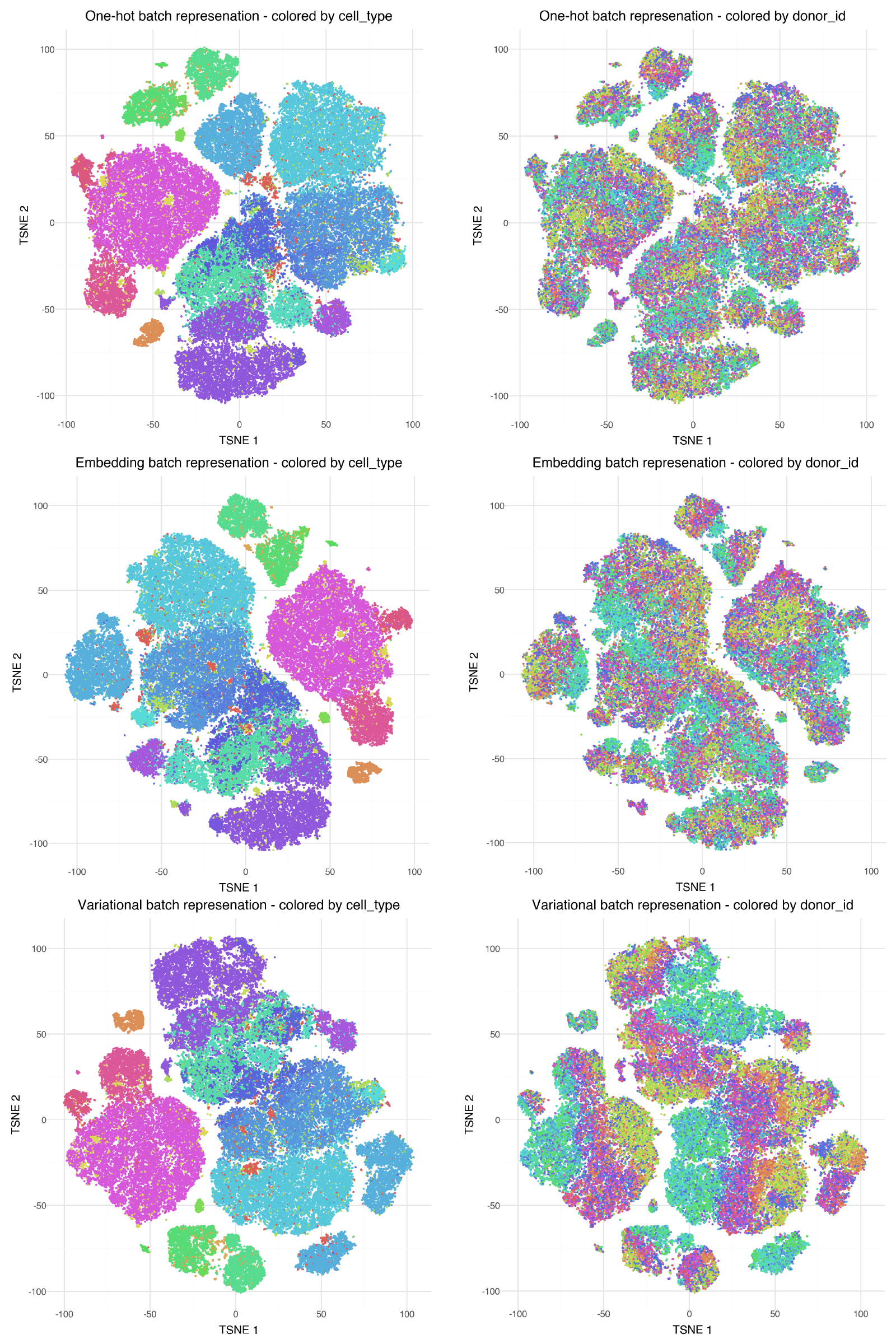
The models were set up to learn cell representations that integrate out difference between donors. The ideal results in this case would show unmixed coloring by cell type (left column), and highly mixed coloring by donor ID (right column). The 'one-hot' and 'embedding' options seem to mix donors about equally well, but the new 'variational' option is not as clearly integrating out variation due to donor.
Variationally encoding unseen batches
The main aim of the new 'varitional' batch representation option is to be able to apply a trained model on new data which has new batches. The model uses a low-dimensional continuous representation of batches. Are these representations meaningful? When we tested the different alternatives above we held out all cells from 50 donors as a test set. Do ‘similar’ unseen donors get variationally encoded to ‘similar’ seen donors?
In the AIDA data, blood samples from donors were processed at three different institutes: the genome institute of Singapore (GIS), RIKEN (RIK), and Samsung genome institute (SGI). We can investigate if donors processed at the different institutes provide similar batch effects by performing TSNE on the 5-dimensional variational batch embeddings representing the donors. In particular, the design of the 'variational' batch representation option also lets us encode donors that were held out from training.
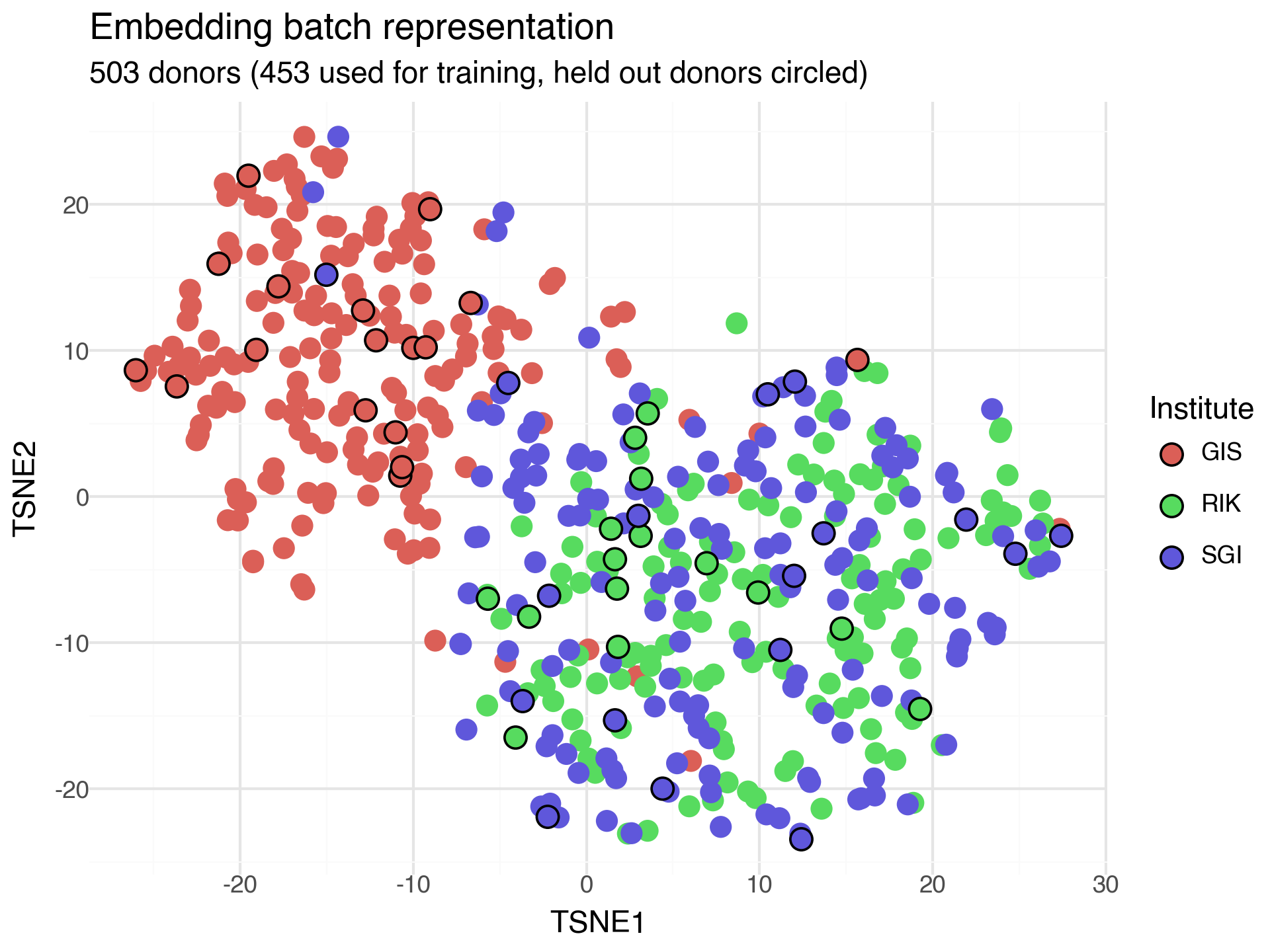
The inference network for the batch representations learns to encode donors from GIS differently from donors from RIK or SGI. In particular, we can see that the batch inference network has generalized, and has successfully encoded unseen donors so they are similar to seen donors from the same institute.
How do the encoded batch embeddings compare to embeddings that are learned through optimization using the 'embedding' batch representation option? We can similarly perform TSNE visualization on these learned embeddings, with the caveat that we can only get these representations for the training data with 453 donors.

Similar to how the 'embedding' option seems better at learning to represent the single cell data, it also appears more effective at learning different types of batch effects. We can see clustering into subpopulations even within institutes in the learned representations.
Conclusions
It is disappointing that this new model using variational encoder for batch representation has lower performance than the other options. It is still very encouraging that the ultimate goal, the ability to represent unseen batches, is working correctly!
It is very likely that 450 training points is insufficient to learn an inference model to encode the batches accurately. It is probably worth revisiting this strategy with a much larger set of batches. There are very few individual datasets with this many batches, but could potentially be useful if training on e.g., all cells in cellxgene and you want to be able to inject new data over time.
A fork and branch of scvi-tools that implements the 'variational' batch representation option is available on github at https://github.com/vals/scVI/tree/codex/add-pseudobulk-generator-and-integrate-with-vae. Scripts and notebooks for training and generating results are available on github at https://github.com/vals/Blog/tree/master/250621-variational-batch-encoding.
References
Tian, Chi, Yuntian Zhang, Yihan Tong, Kian Hong Kock, Donald Yuhui Sim, Fei Liu, Jiaqi Dong, et al. 2024. “Single-Cell RNA Sequencing of Peripheral Blood Links Cell-Type-Specific Regulation of Splicing to Autoimmune and Inflammatory Diseases.” Nature Genetics 56 (12): 2739–52.
From around the web
