
Single-cell metadata as language

Single-cell RNA-seq data is molecule counts of transcripts from different genes within different cells. With this data alone, you can learn which genes tend to covary between cells, or you might learn of groups of cells where different sets of genes are used.
Metadata is data about this data. In the single-cell field this includes information such as which tissue were the cells were harvested from, what technology was used to perform the measurements, or was there some experimental stimulation of the cells.
When you are relating the data and the metadata, you get a connection between the gene expression patterns and external phenomena. You can relate the observations to the rest of the world, either in terms of what observations are related to technical factors, or how observations might change depending on stimuli.Only when you have both gene expression data and the sources of variation in the data can you begin to understand how gene expression is modified in the cells, or the expression in the cells lead to related downstream differences.
When working with single-cell data from multiple sources, you quickly encounter the issue of different people having different ideas on how to format the metadata for the cells. For example, below are the metadata fields and values for two cells from two different data sets.
{
"Index": "CTGAAACAGAATAGGG-11",
"Patient_assignment": "U3",
"Tissue_assignment": "R",
"Disease_assignment": "diseased",
"Celltype": "B"
}
{
"Index": "N52.LPA1b.CCCAGTTTCTGGTTCC-2",
"Cluster": "Plasma",
"Subject": "N52",
"Health": "Non-inflamed",
"Location": "LP",
"Sample": "N52.LPA1b",
"Batch": "2"
}In any analysis of data from the two datasets, the metadata needs to be harmonized. You will need to assign a relation between what parts of data represent the same thing, or different things. Which fields relate to the same concept (e.g., ‘Celltype’ and ‘Cluster’)? Which values within a field represent the same category (e.g., plasma cells are a subset of B cells)?
Once metadata in different datasets are harmonized, and there are subsets of data that are aligned, the different datasets can be put in a model, treated as replicates, or used for comparisons. This metadata harmonization work is equally required no matter the analysis workflow, no matter if it is based on linear models, graph based methods, nonparametric statistics, or deep generative models. The concepts in the metadata need to be aligned to a common vocabulary.
When you are looking at the metadata examples from two cells above, you are probably already aligning the concepts in your head. Clusters and cell types, health status, there appears to be multiple patients and batches which can probably be treated as replicates, etc.
Why can you do this matching? It is because even though the metadata is not standardized, it is text communicating language, in a somewhat structured way. The people who wrote this metadata did actually write it in a way meant to communicate information. They just had slightly different ideas about phrasing. The metadata is language.
At this moment, there are endless language models that can take pieces of text and produce embedding vectors which preserve semantic relations between the texts, in contexts all the way between song lyrics and code in obscure programming languages. These embedding models are designed to preserve that intuition about relations between text which we are using when we can match up the concepts in the metadata examples above.
If we can convert the metadata for the cells to vector representations that preserve the information that the original author meant to communicate, then we can redefine an enormous amount of analysis tasks to circumvent the harmonization step.
A simple version of metadata embedding could be to take the metadata dictionary, convert it to a JSON string, and embed this with a language embedding model, e.g., text-embedding-3-small from OpenAI.
client = OpenAI()
row_ = pd.Series({
"Index": "CTGAAACAGAATAGGG-11",
"Patient_assignment": "U3",
"Tissue_assignment": "R",
"Disease_assignment": "diseased",
"Celltype": "B"
})
js_ = row_.to_json()
response = client.embeddings.create(
input = js_,
model = 'text-embedding-3-small'
)
embeddings = np.array([d.embedding for d in response.data])To investigate this strategy, I went through my collection of scRNA-seq data and sampled 100 random cells from each dataset. I filtered out all cells that didn’t have textual metadata, and was left with 5,760 cells from 58 datasets. I took the textual metadata (that is, I filtered out numeric metadata) for these cells and created 1,536-dimensional embeddings of the JSON representations using the text-embedding-3-small model.
To get some intuition about the embeddings, they can be visualized with PyMDE.

Cell metadata from the same dataset largely group together and do not intermix. There are some cases where we can see the same dataset occupy different regions in the embeddings (for example, purple data in lower right corner).
Do the embeddings represent any interesting information within datasets? As an example, we can zoom in on the first dataset as an example (this is the data with the first dictionary of the first example up top).
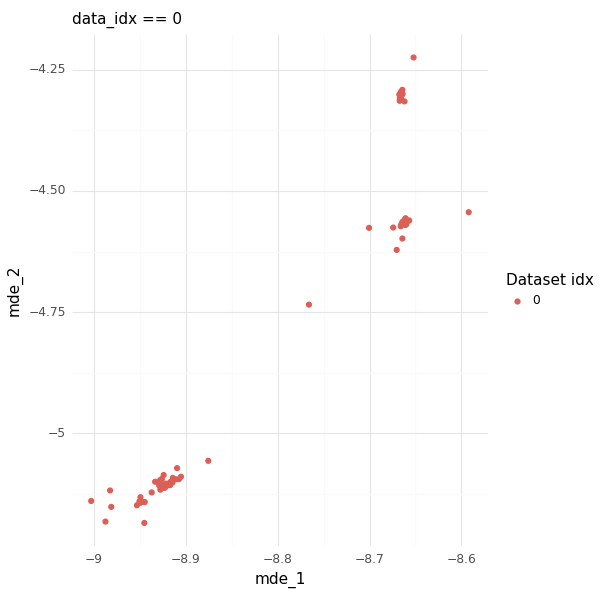
These cells have metadata that cover three distinct regions in the embedding space.

Coloring the cells by some of the metadata information (PBMC vs R [rectum], diseased vs healthy), we can see that the metadata embedding is reflecting that these cells came from different tissues and different disease states.
So, what do these figures indicate? They are pointing to some interesting analysis strategies.
When integrating out variation between datasets, the common choice is a one-hot encoded vector of the length of the number of datasets. Since individual datasets appear to group together in distinct space, the embedding coordinates could be used as continuous covariates in integration models.
Within a dataset, cells with different recorded properties are separated. So also on the local scale, within-dataset, between-condition variation can be modeled with the embedding coordinates as covariates.
This concept can be used with many analysis strategies, but it would tie well with the conditional variational autoencoders in scvi-tools. Zooming in on the relevant part of the SCVI model, a model can be defined with
where s_dataset and s_health end up being one-hot encodings of the dataset and health categories.
Instead, we can imagine creating the cVAE so that it is conditional on the metadata embedding vector (from some function g):
This way, we can tie the gene expression to the author-reported language in the cell metadata. We can analyze the data without spending as much time on metadata harmonization.
There are more potential benefits. We have been thinking about using the language embeddings as a way to represent the observed metadata. Another concept is that the observed language in the metadata can be related directly to language as queries about the data.
The example dataset above mentions two tissue sources for the cells: PBMC, and R. As an example, we can take just the English word “Blood”, and ask how similar the different cells metadata are to this word. Coloring the cells by their metadata distance to “Blood” we see that PBMC-cells are closer than R-cells. We are not informing the model that PBMC is a subset of blood, instead this information is encoded in the OpenAI language model and represented in the embeddings.
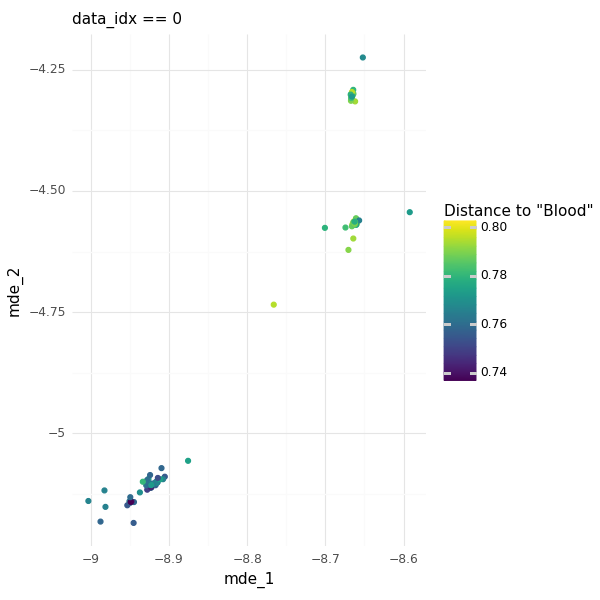
This suggests that if a model is trained with enough single cell data with matched embedded metadata we could get to a point where we can ask differential expression queries using natural language. You can imagine using different natural language queries to filter areas in Z cell transcriptome representation space and define contrasts between areas of metadata embedding space, leading us to highly complex conditional questions.
Here I have just tested a raw embedding of the JSON of the metadata. I couldn’t find any models that had been specifically trained to represent key-value dictionaries. It would be interesting if the language model had distinctions between keys and values encoded in it somehow, though I’m not sure in what way.
Another potential direction could be to fine-tune the language embedding model as the gene expression cVAE model is trained. By iterating between training the cell gene expression representation model and training the metadata embedding model, some gene expression information will inform which terms in the metadata are similar.
A notebook with the analysis presented here is available on GitHub. Thanks to Eduardo Beltrame for helpful feedback on this post.
