Comparing unpublished RNA-Seq gene expression quantifiers
During the conference BoG a new RNA-Seq gene expression quantification tool called Kallisto was announced and released by the Pachter group. This tools have me excited because it is following the trend of Sailfish, RNASkim and recently Salmon. The classical approach for gene (or transcript) expression is aligning reads to genome, then by looking at coverage figuring out which transcripts are expressed at which levels. These new tools in stead use information contained in reads where exact mapping is not completely necessary, and use that to deconvolve the mixture proportions of transcripts in a sample.
To compare these quantifiers, and also using results from a classical TopHat + Cufflinks combination as a control I used five batches of single cell RNA-Seq from three different published data sets: SRP030617 (batch 1), SRP041736 (batch 2 & 3), and EGAS00001001204 (batch 4 & 5). These are all data from human cells, in total 284 samples. The data have between ~100 000 reads and ~5 000 000 reads.
All these data sets have had ERCC spike-ins added to them. To compare quantification accuracy, I first quantify expression for all genes / transcripts plus the spike-ins using the different tools. Then I extracted the TPM values for the spike-ins and looked at the Pearson correlation of the log transformed TPM values and the log transformed concentration of ERCC spike-ins. The plots below shows the distribution of these Pearson R values for the different tools over the different dataset batches. The n refers to the number of samples in a given batch.
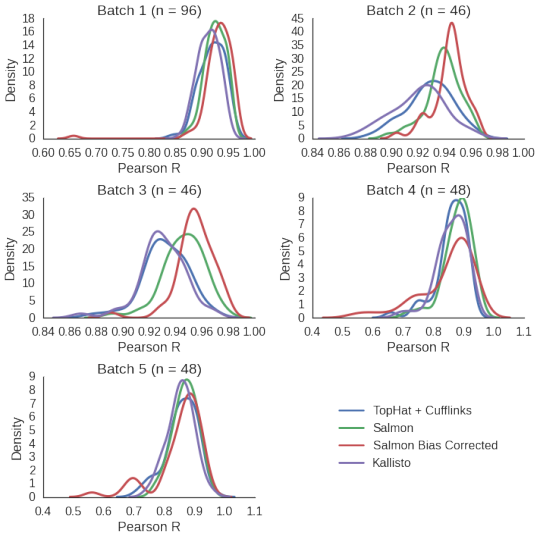
They all perform well, at least equally well to TopHat + Cufflinks, and generally have pretty high correlation values (very few are below 0.8). In some cases Salmon seem to perform better.
To make the task a bit harder, we can look at the input dilution of ERCC’s included in the publications of the data sets and look only at spike-ins present at low levels. In this case given the concentration and dilution in to the volume of the experiments, we look only at the spike-ins present at between 1 and 50 RNA molecule copies.
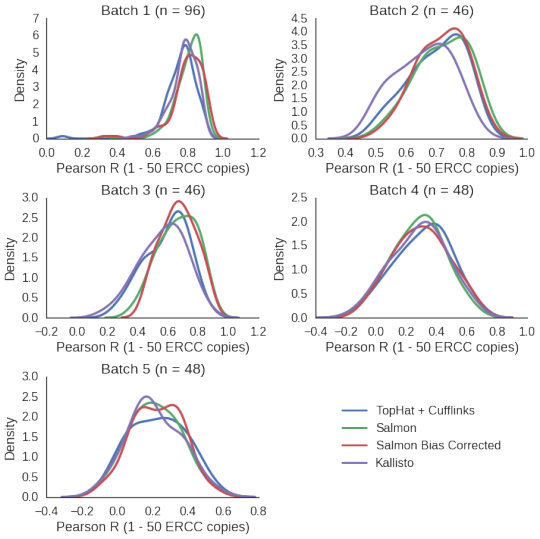
All the methods perform equally, varying from surprisingly good to pretty bad depending on data set.
Regarding speed, the TopHat + Cufflinks combo took something along the lines of a half a day per sample, using multithreading over 12 cores for each sample. (I didn’t save the timings). The Salmon quantification took about 15 minutes per sample, also with 12 cores multithreading, while the Kallisto took about 10 minutes per sample and does not do any multithreading.
It should be noted that since there are no alternative splice forms for the ERCC spike-ins this doesn’t tell us anything about correction or quantification of alternative transcripts.