Immune response enrichment analysis in Python
In a recent paper, Cui and colleagues define a ‘dictionary’ of immune responses to cytokine stimulation (Cui et al. 2023). The experiments performed to produce this dictionary is at an impressive scale, involving injecting 272 mice with 86 different cytokines and harvesting cells from the mice’s draining lymph nodes.
Cytokines are secreted proteins that interact with surface receptors of cells, signaling to the receiving cell to change its current state or perform a function. The immune system heavily relies on this form of signaling.
When a problem occurs, non-immune cells signal to immune cells to activate, to recruit them to the site of the problem, differentiate cells to properly respond to the problem. After the issue is resolved, other cytokines are secreted to decrease the activities of the responding cells. The immune system consists of many components, many different cell types which can handle immune issues in different ways. These components communicate with different sets of cytokines to customize the responses to the problem at hand (Altan-Bonnet and Mukherjee 2019; Saxton, Glassman, and Garcia 2022; Moschen, Tilg, and Raine 2019).
In any diseases or afflictions where the immune system is involved, it is extremely valuable to know which cytokines are involved. The immune dictionary by Cui and colleagues provides, for a large collection of cell type-cytokine pairs, the genes which are transcribed in response to the stimulation from that cytokine. If you know what cell type you are looking at, which genes that cell type has started transcribing, you can look up in the dictionary which cytokine stimulated the cell.
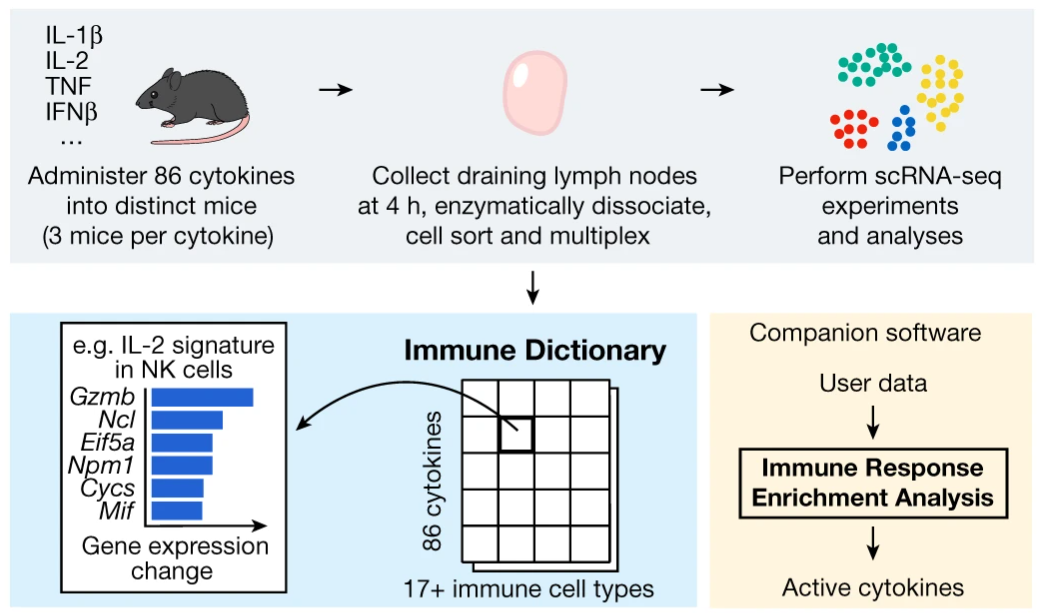
Adapted from https://www.nature.com/articles/s41586-023-06816-9/figures/1
The authors of the immune dictionary created a website at https://www.immune-dictionary.org/ where anyone can upload a list of genes, and infer which cytokines are responsible for the expression of those genes, through what they call ‘immune response enrichment analysis’.
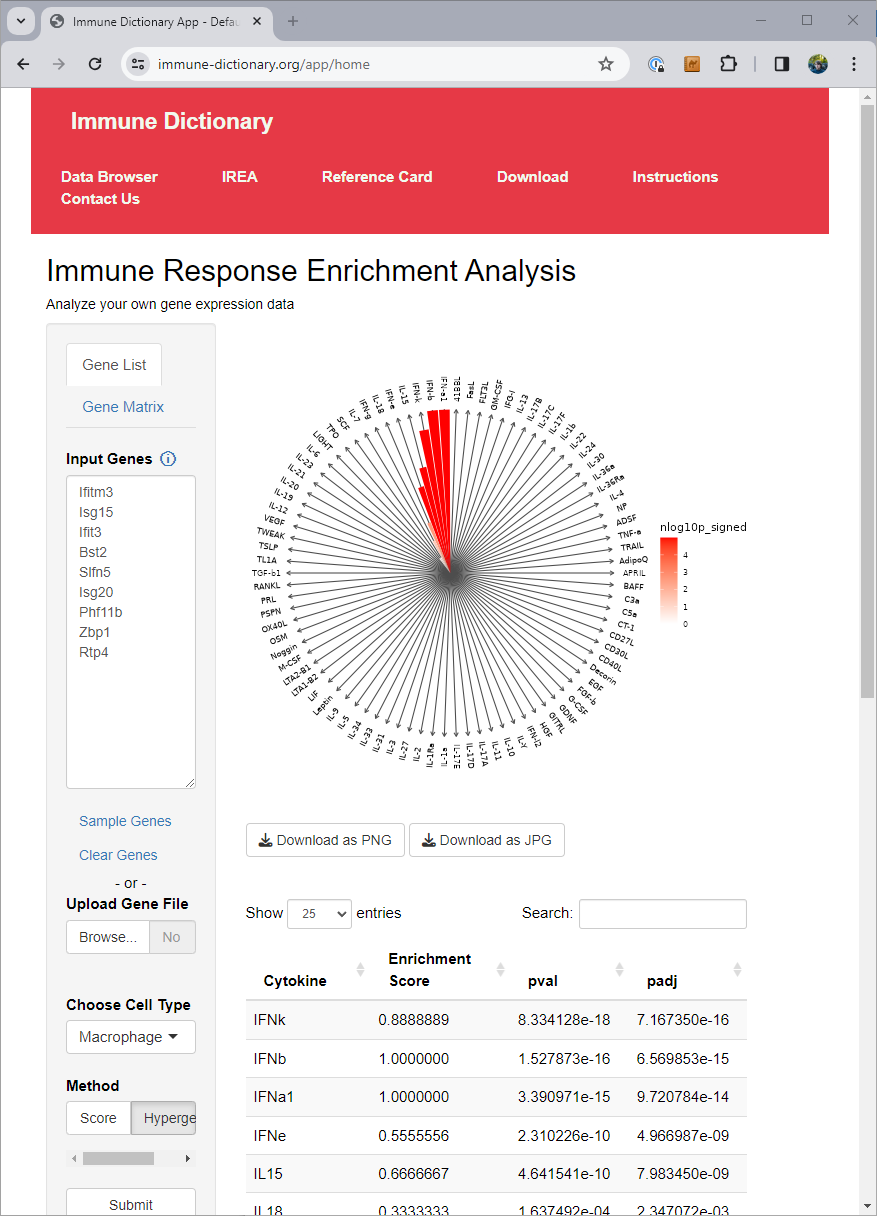
When working in cases with many different conditions or cell types, going to a website and pasting in gene lists for each of them can end up being tedious. At that point you want a solution you can just run as code in your local environment.
The final dictionary from Cui and colleagues, summarizing the results of their large experiment, is available as Supplementary Table 3 in the online version of the paper, having the filename 41586_2023_6816_MOESM5_ESM.xlsx. With these data the tests available on the immune-dictionary.org website can be implemented in Python.
For an example, I found a dataset of mouse colon tissue, with the three conditions of healthy, acute colitis, and chronic colitis, on Figshare. Unfortunately, I haven’t been able to identify what paper these data are from, but it has annotated cell type labels which is very useful for this analysis. By comparing macrophages from healthy mice with macrophages from mice with acute colitis, we can see which genes are activated in the acute colitis case, and compare that list of genes to the genes activated by the different cytokines in the immune dictionary.
After cleaning up the mouse colon data (changing gene identifiers) I ingested it into my Lamin database and fitted an SCVI model to use for differential expression tests.
After reading in the immune response dictionary gene lists from the Excel file, enrichment analysis is done by looking at the overlap between cytokine response genes and genes that are differentially expressed, using, for example, a hypergeometric test.
The Pertpy package’s Enrichment.hypergeometric function would probably be the easiest way to perform the analysis. The gene lists per cytokine stimulation are provided as a dictionary. This function requires your DE results to be stored in a specific format used by scanpy’s rank_genes_groups function.
Since I’m using an SCVI model for differential expression, it will be easier to just obtain a list of DE genes and make use of the code behind the Enrichment.hypergeometric function.
# Differential expression
idx_ = (
vae.adata.obs
.query('celltype_minor == "Macrophage"')
.query('condition in ["Healthy", "AcuteColitis"]')
.index
)
tmp_ = vae.adata[idx_].copy()
de_results = vae.differential_expression(
tmp_,
groupby = 'condition',
group1 = 'AcuteColitis',
group2 = 'Healthy'
)
fde_r = (
de_results
.query('proba_de > 0.95')
)
# Get immune dictionary gene lists
celltype_response = cytokine_responses.query('Celltype_Str == "Macrophage"').copy()
celltype_response['Gene'] = celltype_response['Gene'].map(lambda s: [s])
response_sets = celltype_response.groupby(['Cytokine'])['Gene'].sum().to_dict()
# Calculate hypergeometric gene set enrichment statistics
de_genes = set(fde_r.index)
universe = set(vae.adata.var.index)
results = pd.DataFrame(
1,
index = list(response_sets.keys()),
columns=['intersection', 'response_gene_set', 'de_genes', 'universe', 'pvals']
)
results['de_genes'] = len(de_genes)
results['universe'] = len(universe)
for ind in results.index:
response_gene_set = set(response_sets[ind])
common = response_gene_set.intersection(de_genes)
results.loc[ind, "intersection"] = len(common)
results.loc[ind, "response_gene_set"] = len(response_gene_set)
pval = stats.hypergeom.sf(len(common) - 1, len(universe), len(de_genes), len(response_gene_set))
results.loc[ind, 'pvals'] = pval
results['Cytokine'] = results.indexAfter this we can investigate the results table, or, here, to save some space, visualize the ranking of likely upstream cytokines in a plot.
| | intersection | response_gene_set | de_genes | universe | pvals | Cytokine |
|:-------|---------------:|--------------------:|-----------:|-----------:|-----------:|:-----------|
| IL-21 | 1 | 1 | 96 | 18416 | 0.00521286 | IL-21 |
| RANKL | 1 | 1 | 96 | 18416 | 0.00521286 | RANKL |
| IL-13 | 2 | 34 | 96 | 18416 | 0.0135363 | IL-13 |
| IL-17F | 1 | 3 | 96 | 18416 | 0.015558 | IL-17F |
| OSM | 1 | 7 | 96 | 18416 | 0.0359301 | OSM |
...We see that, based on the immune dictionary, the macrophages in the colon of mice with acute colitis are likely to have been stimulated by the cytokines IL-21, RANKL, IL-17F, IL-13, and OSM.
The notebooks used for this post are available on GitHub, the data used for this post are available on LaminDB, and the trained SCVI model is available from Hugging Face using scvi-hub. Thanks to Eduardo Beltrame for helpful feedback on this post.
References
Altan-Bonnet, Grégoire, and Ratnadeep Mukherjee. 2019. “Cytokine-Mediated Communication: A Quantitative Appraisal of Immune Complexity.” Nature Reviews. Immunology 19 (4): 205–17. https://doi.org/10.1038/s41577-019-0131-x.
Cui, Ang, Teddy Huang, Shuqiang Li, Aileen Ma, Jorge L. Pérez, Chris Sander, Derin B. Keskin, Catherine J. Wu, Ernest Fraenkel, and Nir Hacohen. 2023. “Dictionary of Immune Responses to Cytokines at Single-Cell Resolution.” Nature, December. https://doi.org/10.1038/s41586-023-06816-9.
Moschen, Alexander R., Herbert Tilg, and Tim Raine. 2019. “IL-12, IL-23 and IL-17 in IBD: Immunobiology and Therapeutic Targeting.” Nature Reviews. Gastroenterology & Hepatology 16 (3): 185–96. https://doi.org/10.1038/s41575-018-0084-8.
Saxton, Robert A., Caleb R. Glassman, and K. Christopher Garcia. 2022. “Emerging Principles of Cytokine Pharmacology and Therapeutics.” Nature Reviews. Drug Discovery, September. https://doi.org/10.1038/s41573-022-00557-6.


