
Low mapping rate 6 - Converting sorted BAM to FASTQ

Some sequencing centers have moved to only work on BAM or CRAM files rather than "raw" FASTQ files. The motivation for this is that CRAM files can be heavily compressed and require less memory for the sequencing service provider. The CRAM files in particular make use of the alignment to a reference genome to achieve better compression performance, especially when the files are sorted by alignment coordinate.
In many forms of sequencing analysis, in particular genetics, coordinate sorted alignments to a standard reference is so standardized it is considered a "raw data" format. Unfortunately, this is not the case for RNA sequencing, in single cells or other. As demonstrated by the previous posts in the series, we are often not even sure what material we have been sequencing. The choice of what cDNA's are in the reference is also an informed choice depending on the experiment. For these reasons, software for working with RNA-seq data require FASTQ's as input.
To convert BAM's/CRAM's to FASTQ, a handy command from the samtools suite is samtools fastq. It is however not mentioned in the documentation or in the help for the command itself that is assumes name sorted BAM/CRAM input. It will not even stop or warn you if your file is coordinate sorted. Instead, it will silently create paired FASTQ files with incorrect read orders.
This issue was recently raised by Davis on twitter.
Is it common knowledge that CRAM files (may) need to be sorted before conversion to fastq?
e.g.
samtools sort -m 10G -n -T %s {input} | samtools fastq -F 0xB00 -1 {out.fq1} -2 {out.fq2} -'
may give different results to
samtools fastq -t -1 {out.fq1} -2 {out.fq2} {input}'— Davis McCarthy (@davisjmcc) November 16, 2017
In particular, the incorrectly generated FASTQ files will have worse performance in terms of mapping statistics, caused by read pairs not originating from the same cDNA fragment. We can compare the result on the same data and reference as we used before, after converting files with name sorted and coordinated sorted CRAM files.
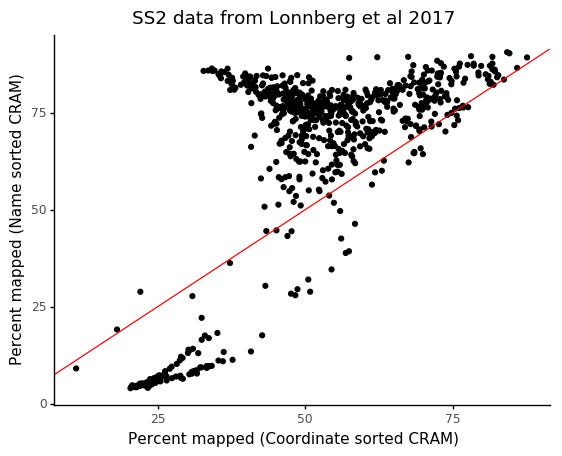
We see that cells which would have a mapping rate of > 75% only have ~40% mapping rate with the incorrect coordinate sorting.
Thomas Keane recommends using the samtools collate command before converting to FASTQ to quickly ensure reads are correctly ordered.
SAM/BAM/CRAM->fastq: samtools view | samtools collate | samtools fastq. Collate is quicker than sort as it does local grouping of alignments by name. Agree should be in docs, send pull request.
— Thomas Keane (@drtkeane) November 17, 2017
If you have data with lower mapping rates and you were provided BAM or CRAM files by your facility, it might be worth to check the sort order. This can be seen in the first line of a BAM/CRAM file.
$ samtools view -H 20003_4#70.cram | head | cut -c1-50
@HD VN:1.4 SO:coordinate
@RG ID:20003_4#70 DT:2016-06-07T00:00:00+0100 PU:1
@PG ID:SCS VN:2.2.68 PN:HiSeq Control Software DS:
@PG ID:basecalling PP:SCS VN:1.18.66.3 PN:RTA DS:B
@PG ID:Illumina2bam PP:basecalling VN:V1.19 CL:uk.
@PG ID:bamadapterfind PP:Illumina2bam VN:2.0.44 CL
@PG ID:BamIndexDecoder PP:bamadapterfind VN:V1.19
@PG ID:spf PP:BamIndexDecoder VN:v10.26-dirty CL:/
@PG ID:bwa PP:spf VN:0.5.10-tpx PN:bwa
@PG ID:BamMerger PP:bwa VN:V1.19 CL:uk.ac.sanger.nSimilar to the case of unsorted FASTQ files, you can also notice this by read headers not matching up in the FASTQ pairs.
$ head 20003_4#70_*.fastq | cut -c1-50
==> 20003_4#70_1.fastq <==
@HS36_20003:4:2214:5946:43267
CTATTAAGACTCGCTTTCGCAACGCCTCCCCTATTCGGATAAGCTCCCCA
+
BBBBBBBFFFFFF/FFFFFF/<F</FFF/B/<<F/FFF/<<F<B/F/FFF
@HS36_20003:4:2111:19490:55868
GTATCAACGCAGAGTACTTATTTTTTTTTTTTTTTTTTTTTTTGGGTGGT
+
BBBBBFFFBFFFFFFFFFF/<<<FBBBFFFFFFFFFFFFFFFB///////
@HS36_20003:4:2109:19213:11514
TATCAACGCAGAGTACATGGGTGGCGCCAGCTTCAGCAAAAGCCTTTGCT
==> 20003_4#70_2.fastq <==
@HS36_20003:4:2214:5946:43267
GCCCTTAAGTTGATTTGAGAGTAGCGGAACGCTCTGGAAAGTGCGGCCAT
+
BBBBBBFFBFFFF/FFBFFFB/<FFFFF<FFFBFFFFFBFFFFFFFFF<F
@HS36_20003:4:1111:7773:69672
GAGACATAATAAACAAAAAAAGAATAAGATAAAGGAGAGAGAAAAAAACG
+
/<//////<//BF/F/FFFFF/<F/B//</<FF//F/F/F/FFFFFFF</
@HS36_20003:4:2109:19213:11514
CATTACAGGCGCATCTTACGGAATTGGATATGAAATAGCAAAGGCTTTTGThis post was suggested by Raghd Rostom and Davis McCarthy.
