Detecting scRNA-seq study duplicates using sentence embeddings
I have been maintaining a spreadsheet of publications that generated single-cell transcriptomics data for about five years. It is linked at the top of this website, and we wrote up a paper about it a few years ago (Svensson, da Veiga Beltrame, and Pachter 2020). Earlier today it had 1,933 entries.
Naturally, over time, mistakes such as inserting the same publication twice are bound to happen. I took some time today to identify accidental duplicates.
My initial approach was to search for entries with exactly the same DOI (digital object identifier; the unique string that identifies a publication). I found four papers with this strategy and deduplicated them.
In the spreadsheet, a DOI is the only required field, from which an author list, a title, a publication, and a date are collected automatically with a macro that calls the CrossRef API. Even though DOIs are unique identifiers of papers, sometimes the same study is duplicated with different DOIs. The main reason for this is that a paper gets a DOI when it is posted on a preprint server such as bioRxiv, and then a second DOI once it is published in a peer reviewed journal.
To identify preprint-journal duplicates, my first strategy was to find papers with exactly the same title. This identified five studies, all of which turned out to be the same but one being on bioRxiv and one being in a journal.
A paper that has been revised before being submitted to a journal, then further revised through the review process, and finally has to adhere to the style guide of the journal, is unlikely to retain exactly the same title. To find papers that were likely duplicates, I needed a way to identify titles that probably describe the same paper even if there are slight variations.
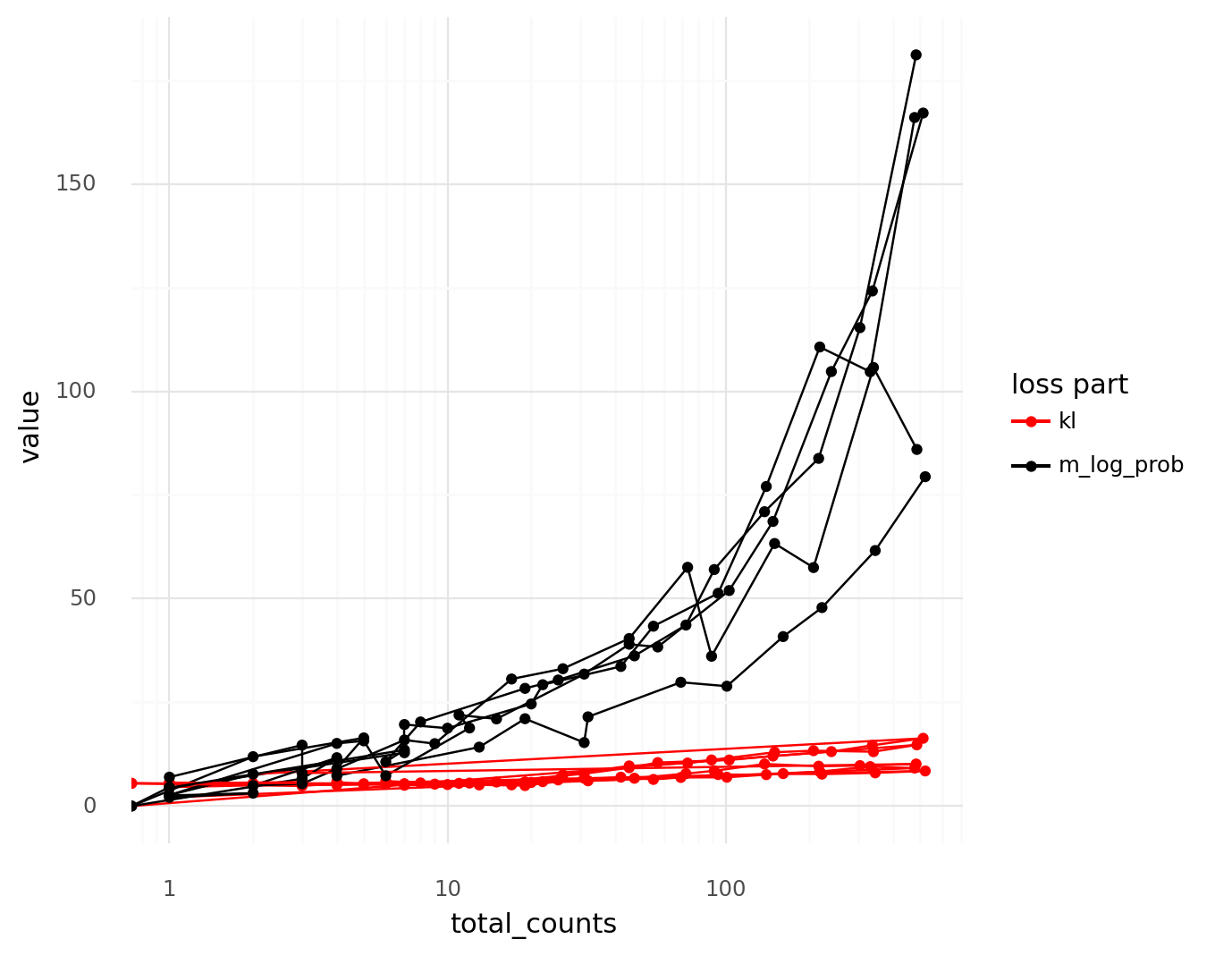
The quickest and simplest approach I came up with was to use the OpenAI API to create sentence embeddings of the titles. Then calculate all the pairwise distances between the embeddings, and look at the pairs of titles that were the closest to each other. This ended up being very simple and effective!
client = OpenAI()
responses = []
for chunk in tqdm(np.array_split(data, 10)):
query = chunk['Title'].to_list()
response = client.embeddings.create(input = query, model = 'text-embedding-ada-002')
responses += [response]
embeddings_list = []
for response in responses:
embeddings = np.array([d.embedding for d in response.data])
embeddings_list += [embeddings]
embeddings = np.vstack(embeddings_list)
pdists = sklearn.metrics.pairwise_distances(embeddings)
mask = np.triu(np.ones(pdists.shape), k = 1).astype(bool)
pdistsl = pd.DataFrame(pdists).where(mask).stack().reset_index()
top_similar = pdistsl.sort_values(0).head(20)
for _, r in top_similar.iterrows():
print('Distance:', r[0])
d_r_0 = data.iloc[r['level_0'].astype(int)]
d_r_1 = data.iloc[r['level_1'].astype(int)]
print(d_r_0['DOI'], '\n|', d_r_0['Title'])
print(d_r_1['DOI'], '\n|', d_r_1['Title'])
print()
Distance: 0.04542879727657215
10.1101/2020.10.07.329839
| Single-nucleus transcriptome analysis reveals cell type-specific molecular signatures across reward circuitry in the human brain
10.1016/j.neuron.2021.09.001
| Single-nucleus transcriptome analysis reveals cell-type-specific molecular signatures across reward circuitry in the human brain
Distance: 0.051210665464434646
10.1101/2020.07.11.193458
| Single-nucleus RNA-seq2 reveals a functional crosstalk between liver zonation and ploidy
10.1038/s41467-021-24543-5
| Single-nucleus RNA-seq2 reveals functional crosstalk between liver zonation and ploidy
Distance: 0.07003401486501365
10.1101/2020.03.02.955757
| Diversification of molecularly defined myenteric neuron classes revealed by single cell RNA-sequencing
10.1038/s41593-020-00736-x
| Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing
Distance: 0.08156853865981699
10.1101/2021.07.19.452956
| The Tabula Sapiens: a multiple organ single cell transcriptomic atlas of humans
10.1126/science.abl4896
| The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans
Distance: 0.1182708273417854
10.1101/2020.04.22.056341
| Deconvolution of Cell Type-Specific Drug Responses in Human Tumor Tissue with Single-Cell RNA-seq
10.1186/s13073-021-00894-y
| Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq
Distance: 0.14183682263019862
10.1101/2020.01.19.911701
| Surveying Brain Tumor Heterogeneity by Single-Cell RNA Sequencing of Multi-sector Biopsies
10.1093/nsr/nwaa099
| Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies
Distance: 0.15672052837461234
10.21203/rs.3.rs-745435/v1
| Single cell analysis of endometriosis reveals a coordinated transcriptional program driving immunotolerance and angiogenesis across eutopic and ectopic tissues.
10.1038/s41556-022-00961-5
| Single-cell analysis of endometriosis reveals a coordinated transcriptional programme driving immunotolerance and angiogenesis across eutopic and ectopic tissues
Distance: 0.16437164718666886
10.1101/2020.06.17.156943
| Chromatin potential identified by shared single cell profiling of RNA and chromatin
10.1016/j.cell.2020.09.056
| Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin
Distance: 0.16911884570096825
10.1101/2021.04.24.441206
| Single-cell landscapes of primary glioblastomas and matched organoids and cell lines reveal variable retention of inter- and intra-tumor heterogeneity
10.1016/j.ccell.2022.02.016
| Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity
Distance: 0.183893761793663
10.1101/2020.02.12.946509
| No detectable alloreactive transcriptional responses during donor-multiplexed single-cell RNA sequencing of peripheral blood mononuclear cells
10.1186/s12915-020-00941-x
| No detectable alloreactive transcriptional responses under standard sample preparation conditions during donor-multiplexed single-cell RNA sequencing of peripheral blood mononuclear cells
Distance: 0.18895108556159476
10.1101/2020.01.13.891630
| Single-cell transcriptome analysis reveals cell-cell communication and thyrocyte diversity in the zebrafish thyroid gland
10.15252/embr.202050612
| Single‐cell transcriptome analysis reveals thyrocyte diversity in the zebrafish thyroid gland
Distance: 0.2003776161396695
10.21203/rs.3.rs-599203/v1
| A Single-cell Interactome of Human Tooth Germ Elucidates Signaling Networks Regulating Dental Development
10.1186/s13578-021-00691-5
| A single-cell interactome of human tooth germ from growing third molar elucidates signaling networks regulating dental development
Distance: 0.23986737520644938
10.1101/2022.01.12.476082
| Scalable in situ single-cell profiling by electrophoretic capture of mRNA
10.1038/s41587-022-01455-3
| Scalable in situ single-cell profiling by electrophoretic capture of mRNA using EEL FISH
Distance: 0.25840869095237246
10.2337/db16-0405
| Single-Cell Transcriptomics of the Human Endocrine Pancreas
10.1016/j.cels.2016.09.002
| A Single-Cell Transcriptome Atlas of the Human Pancreas
Distance: 0.26278269347286093
10.15252/embj.2018100811
| A single‐cell transcriptome atlas of the adult human retina
10.1093/nsr/nwaa179
| A single-cell transcriptome atlas of the aging human and macaque retina
Distance: 0.26422422020526076
10.1038/s41467-018-08079-9
| Single-cell transcriptomic analysis of mouse neocortical development
10.1101/2020.04.23.056390
| Single-cell transcriptomic analysis identifies neocortical developmental differences between human and mouse
Distance: 0.2916387113759244
10.1038/s41586-022-04518-2
| A single-cell atlas of human and mouse white adipose tissue
10.1038/s41467-023-36983-2
| An integrated single cell and spatial transcriptomic map of human white adipose tissue
Distance: 0.29364709869497707
10.1016/j.devcel.2020.05.010
| Single-Cell RNA Sequencing of Human, Macaque, and Mouse Testes Uncovers Conserved and Divergent Features of Mammalian Spermatogenesis
10.1016/j.devcel.2020.07.018
| Single-Cell RNA Sequencing of the Cynomolgus Macaque Testis Reveals Conserved Transcriptional Profiles during Mammalian Spermatogenesis
Distance: 0.2940628150528307
10.1126/science.aar4362
| Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo
10.1101/2021.10.21.465298
| Spatiotemporal mapping of gene expression landscapes and developmental trajectories during zebrafish embryogenesis
Distance: 0.2991743187594748
10.1101/2022.02.01.478648
| Single-cell RNA profiling of Plasmodium vivax liver stages reveals parasite- and host-specific transcriptomic signatures and drug targets
10.1371/journal.pntd.0010633
| Single-cell RNA sequencing of Plasmodium vivax sporozoites reveals stage- and species-specific transcriptomic signatures
The majority of highly similar article titles were bioRxiv preprints with their matched journal publications, and a couple of medRxiv and ResearchSquare preprints. Some were genuinely different papers that just happened to have very similar titles. Through this process, I could remove 14 more duplicates. In total, I discovered 23 paper duplicates!
I was particularly impressed with how easy it is to get high quality sentence embeddings at this time. There are probably technically simpler strategies to get similar titles, such as removing punctuation and converting all characters to lowercase, both of which mostly depend on journal style guides. But in actuality, at this point, just getting text embeddings will be easier than any ad hoc strategy.
A notebook with code related to this post is available at Github
References
Svensson, Valentine, Eduardo da Veiga Beltrame, and Lior Pachter. 2020. “A Curated Database Reveals Trends in Single-Cell Transcriptomics.” Database: The Journal of Biological Databases and Curation 2020 (November). https://doi.org/10.1093/database/baaa073.